
OBJECTIVE:
To lay down a procedure for the validation process in terms of evaluation, planning, qualification, re-qualification and management of validation documentation.
SCOPE:
This SOP is applicable to the validation and qualification of process facilities, utilities, equipment, analytical instruments, and computerized systems for use according to cGMP. Herein “System/Systems” will be used interchangeably as the collective name for process facilities, utilities, equipment, analytical instruments and computerized systems. In the context of GLP operations, this procedure applies only to computerized systems. This SOP is applicable to {COMPANY NAME} {COMPANY LOCATION}.
RESPONSIBILITY:
Validation Team/Project Sponsor/Owner (herein referred to as the User)
Users are responsible for ensuring that:
All Systems are validated in accordance with this procedure.
Validation documentation is prepared, reviewed, approved, reported and archived prior to utilization of the System for routine GMP/GLP operations.
They review and approve all validation documentation generated in support of the systems.
Signature definition: Validation has been carried out and documented according to the Validation Procedure SOP and all deviations have been recorded and resolved.
Technical Support
Depending on the type of the Qualification/Calibration, Technical Support may be provided by:
Technical experts within Organization (specialized systems, analytical instruments).
Engineering (facilities, utilities and equipment)
Information Systems (computerized systems)
Specialized system (A supplier).
A combination of the above (this should be decided during the Evaluation and Planning phase where the project/validation team is assembled).
The Technical support is responsible for delivering Validation Systems (i.e. ensuring that the design, construction, testing, installation and commissioning of Systems are adequately documented and controlled). Technical Support will review and approve validation documentation for Systems.
ACCOUNTABILITY:
QA Head shall be accountable for Approval, Training, Implementation and Execution of this SOP
PROCEDURE:
DEFINITIONS & TERMINOLOGY:
Protocol: A Protocol is a written set of instructions broader in scope than a Standard Operating Procedure (SOP).
System: Referred in this SOP means Facility / Equipment / Instrument or Utility.
Activity: Any monitoring or measuring or Qualification step.
Validation / Qualification: Documented act of proving that any Facility / Utility / Equipment / Instrument / System actually lead to expected results.
URS (User Requirement Specification): Document indicating User Requirements and Specifications of any Equipment / Utility / System, submitted to Vendor(s) to ensure that the Equipment / Utility / System meet the intended purpose.
Design Qualification (DQ): Documented verification that the proposed design of the Facilities, Systems and Equipment is suitable for intended purpose.
Installation Qualification (IQ): Documented verification that the Facilities, Systems and Equipment as installed or modified, comply with the approved design & the Manufacturer’s recommendation.
Operational Qualification (OQ): Documented verification that the Facilities, Systems and Equipment as installed or modified, perform as intended throughout the anticipated Operating Range & is operating consistently as per the established specification or design criteria.
Performance Qualification (PQ): Documented verification that the Facilities, Systems and Equipment as connected together, can perform effectively and reproducibly, to perform approved process and deliver product specification consistently.
Re-Qualification: Planned, periodic Qualification to prove that any Facility / Utility / Equipment/ Instrument / System actually are in validated state.
Modification: Any significant change which may alter the validated state of Facility / Utility / Equipment /Instrument / System.
Relocation: Change in location of any Utility, Equipment or System.
GENERAL GUIDELINES FOR PREPARATION & APPROVAL OF QUALIFICATION, VALIDATION PROTOCOL & REPORTS:
Protocols & Reports shall be written in English Language by using Microsoft Word typing.
Protocols & Reports shall be initiated by User Department / QA Department.
The person directly supervising the Process or Activity shall initiate New Protocols & Reports & Revision of Protocols & Reports.
Protocols & Reports shall be written after thorough understanding of the Process & Activity. The Initiator of the Protocols & Reports shall have adequate knowledge, Training and experience in the related Areas of activity.
All the points in the Protocol / Report shall be numbered sequentially and sub paragraph of the Protocol / Report be also numbered sequentially with an incremental number derived from the heading number. Bullets May be use for sub paragraph of Protocol / Report.
The User Requirement Specification (URS), Design Specification (DS) and Functional Specification (FS) shall be required for Tailor Made System only. For Standard System (like Standard Weighing Balance & HPLC, etc), URS is not essential, only Design, Installation, Operational & Performance Qualification (as applicable) shall be done.
Initiator shall check the completeness of draft Protocols & Reports and send the hardcopy to the Head of the Department for review.
The reviewer shall check the draft Protocols & Reports for accuracy of the subject matter, Logical Sequence of Process. On receipt of the comments (if any), in the Protocols & Reports if agreed.
Click the link for download word file copy of this document:
https://pharmaguidehub.com/product/standard-operating-procedure-on-validation-and-qualification-procedure-for-facility-utility-equipment-and-process/
Protocols & Reports shall have reference of related document such as Pharmacopoeia and Guidelines published by various Regulatory Authorities. Wherever necessary illustrations and drawing shall be indicated to provide better clarity and understanding of the Process / System.
The draft Protocols & Reports after completion of checking shall be destroyed; this shall be done in concerned Originating Department.
The Soft Copy of final Protocols & Reports shall be transferred to QA Department and the soft copy in other Departments shall be deleted in presence of QA person.
Master Copies of all Protocols & Reports shall be printed in QA department.
Protocols & Reports should be handled with care and should not be spoiled or torn. & No manual correction shall be made in Approved Copy.
All Master Copy / Soft Copy of Approved Protocols & Reports shall be stored in QA Department and Data Backup shall be kept in Information Technology (IT) Department.
All Protocols & Reports shall be prepared by Operating Officer / Executive of concerned Department, Reviewed by Head of the concerned Department / Head of related activity/Designe & Approved by Head Quality Assurance.
Note:
Irrespective of given Annexures of all protocol and reports, Headings and Sub headings shall be modified, new heading or sub heading shall be added and existing heading shall be deleted as per requirements.
Protocol / Report/Pre/post Approval
INITIATED BY
| DESIGNATION | NAME | SIGNATURE | DATE |
| OFFICER / EXECUTIVE (DEPARTMENT) | – | – | – |
REVIEWED BY
| DESIGNATION | NAME | SIGNATURE | DATE |
| HEAD (DEPARTMENT) | – | – | – |
| HEAD (ENGINEERING) | – | – | – |
| MANAGER / DESIGNEE (QUALITY ASSURANCE) | – | – | – |
APPROVED BY
| DESIGNATION | NAME | SIGNATURE | DATE |
| HEAD (QUALITY ASSURANCE) | – | – | – |
Protocols & Reports should have a unique Reference Number, which shall identify that document. The document number shall be given as per point No. 5.7 (Numbering system of protocol and report)
Content of Protocol/Report: content of protocol/report in the form of given table. Header Row of Table shall have gray 25 % Shading Colour.
PROTOCOL/REPORT CONTENTS:
| S. No. | TITLE | PAGE NO. |
| – | – | – |
Font size of table Content may be Changeable in case of insufficient space but it should be not less than 9 Font.
Depending upon System, Utility / Area the content of Protocol/Report may vary.
After preparation & approval of Protocol, Photocopy of Master Copy (as Controlled Copy) shall be used for Execution of Qualification / Validation activity.
After completion of Qualification / Validation, a Qualification / Validation Report shall compile. The Protocol / Report shall be reviewed by User Department i.e. QC Head / Engineering Head/ Production Head (as applicable), Approved by QA Head.
The approval of Qualification / Validation Report shall consider as handover of the system to user Department for its intended purpose.
Qualification Protocol shall be separate for DQ, IQ, OQ, & PQ for each activity & report also separate.
The activity flow for Qualification and Validation process is as:
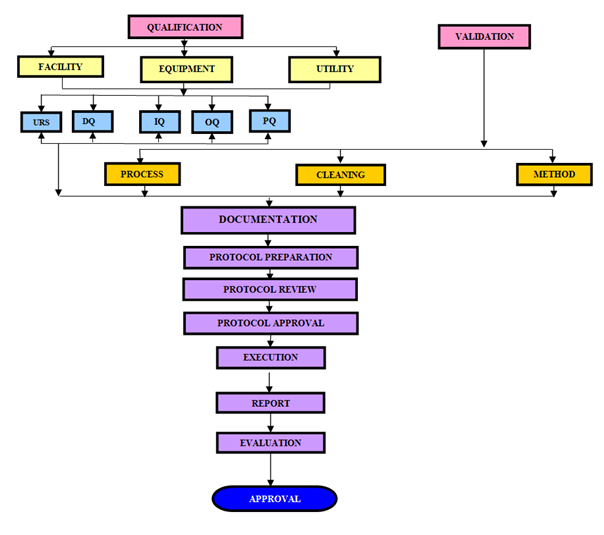
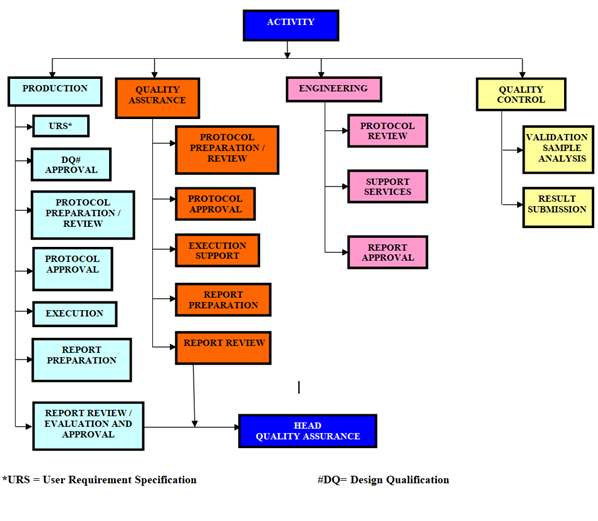
The Responsibility & activity flow for Qualification and Validation process is as:
Content of Header and Footer:
Header:
The Header of Qualification/Validation documents shall have the Name of Organization (Including Name of Location). Header shall have the “Logo” of Organization in Left corner on Top and Title for Example “User Requirement Specification for———–” in centerwritten in Bold and Capital letter of font size 12 as shown in Annexure-VI.
Document no. Shall be written as per below next mentioned steps
Effective Date: Effective Date shall be assigned in the form of DD/MM/YY.Shall be written in bold and Capital Letter of Font Size 12. Shall be Pre-printed.
Page No.: Shall be written in Bold and Capital of Font Size 12. The Page Number shall be mentioned in ‘X of Y’ format. For Example: If a URS contains 60 pages, then the first page of the URS shall be 1 of 60 and the second page shall be 2 of 60 respectively. Shall be Pre-printed.
Footer:
Format No.: 09 Normal & Capital font size printed on the Left Corner of the page after Footer, out of page border and shall be printed as for Example: QA-003/F05-00 on all pages of the Protocol.
Click the link for download word file copy of this document:
https://pharmaguidehub.com/product/standard-operating-procedure-on-validation-and-qualification-procedure-for-facility-utility-equipment-and-process/
PREPARATION OF QUALIFICATION PROTOCOL AND REPORTS:
Note:
DQ, IQ, OQ and PQ are provided by vendor as per their format.
If not provided than in house DQ, IQ, OQ and PQ shall be prepared based on URS and information provided by vendor.
Preparation of Protocol shall be initiated after receipt of approved Specification and Design.
NUMBERING SYSTEM OF QUALIFICATION PROTOCOLS & REPORTS:
Each Protocols & Reports shall have unique No. for Identification and Control. Once a number is assigned to any Protocol & Report, the same number shall not be assigned to any other Protocols & Reports.
The Qualification Protocol and report (OQ, IQ, PQ) No. shall be assigned as:
For IQ the numbering system shall be assign as: WWW/XX/YYY/ZZZ
Where,
WWW – Indicate Qualification protocol/Report name e.g. (Installation Qualification Protocol/Report IQP/IQR)
XX- Indicate Machine Location e.g. (Production, QC/Micro)
YYY- Indicate Equipment Name e.g. (Tablet Compression Machine (TCM)) ZZZ – Indicate Equipment ID serial No. start from 001, 002, 003,004…………………
E.g., IQP/PD/TCM/001 and Report No. IQR/PD/TCM/001
For OQ the numbering system shall be assign as: WWW/XX/YYY/ZZZ
Where,
WWW – Indicate Qualification protocol/Report name e.g. (Operational Qualification Protocol/Report OQP/OQR)
XX- Indicate Machine Location e.g. (Production, QC/Micro)
YYY- Indicate Equipment Name e.g. (Tablet Compression Machine (TCM))
ZZZ – Indicate Equipment ID serial No. start from 001, 002, 003,004…………………
E.g. OQP/PD/TCM/001 and Report No. OQR/PD/TCM/001
For PQ the numbering system shall be assign as: WWW/XX/YYY/ZZZ
Where,
WWW – Indicate Qualification protocol/Report name e.g. (Performance Qualification Protocol/Report PQP/PQR)
XX- Indicate Machine Location e.g. (Production, QC/Micro)
YYY- Indicate Equipment Name e.g. (Tablet Compression Machine (TCM))
ZZZ – Indicate Equipment ID serial No. start from 001, 002, 003,004…………………
E.g. PQP/PD/TCM/001 and Report No. PQR/PD/TCM/001
AREA QUALIFICATION PROTOCOL AND REPORT
Area Qualification protocol and report shall be prepared by engineering person.
The numbering system for area qualification protocol and report as given below
XXX/YYY
Where,
XXX: Indicate Area Qualification protocol/Report
YYY: Indicate Serial No. start from 001, 002, 003……………….
E.g. AQP/001, AQR/001
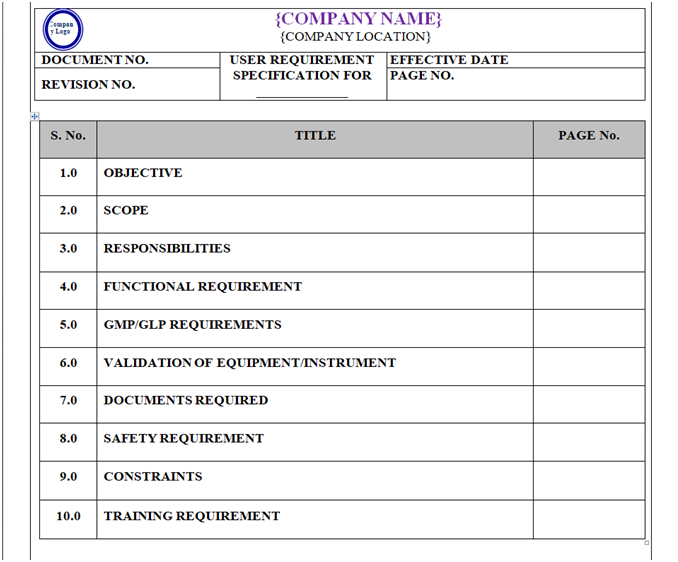
USER REQUIREMENT SPECIFICATION (URS) –(As applicable):
The numbering system for user specification requirement shall be assigned as:
URS/XXX/YYY
URS: indicate user specification requirement
XXX: Equipment Name
YYY: Equipment ID No. start from 001, 002, 003……………
E.g. URS/AHU/001
Content of Header and Footer: refer point no. 5.2.24.
Content of Body Part:
URS shall contain following sections / Heads but not limited to:
- Objective
- Scope
- Responsibility
- Functional Requirements
- GMP / GLP Requirements
- Validation of Equipment/Instrument
- Document Required
- Safety Requirement
- Constraints
- Training Requirement
- Discussion, Review and comments
- Abbreviation
- Approval Sheet
- Revision History
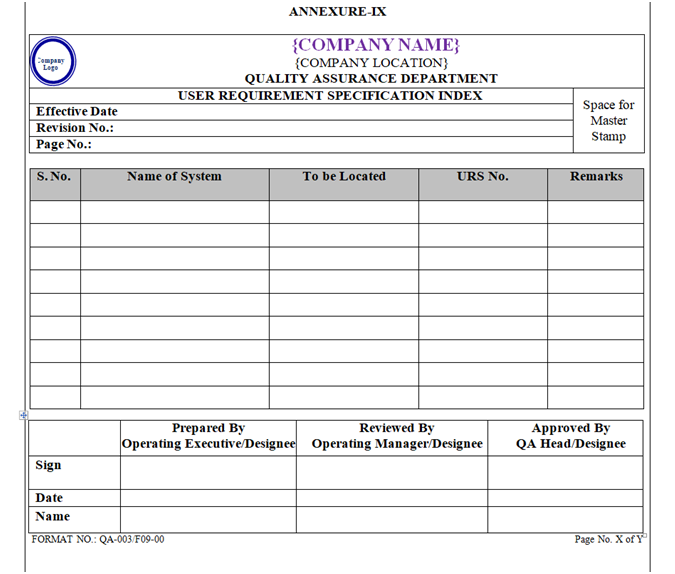
The URS shall be numbered serially and the list of the same shall be maintained by Quality Assurance Department as per Annexure-IX &shall be updated Bi-annually or whenever required.
The URS shall be initiated by officer / Executive of user Department, Reviewed by Head of User Department/Designee i.e. QC / Engineering/ Production / Warehouse (which ever is applicable), Approved by QA Head/Designee.
A copy of approved URS shall be forwarded to vendor(s).
If there is any modification in the URS, the same shall be revised and the revised copy of the same shall be sent to the vendor(s).
On the basis of URS, the vendor(s) shall send the Design Specification (DS) and / or Functional Specification (FS).
The DS / FS received from the vendor(s), if found acceptable shall be approved by Head of User Department / QC / Engineering / Head Production and QA.
The Design of the system thus Qualified and Approved, is considered as Design Qualification (DQ). DQ shall be communicated to Vendor / Supplier.
Any change in the system after DQ shall be incorporated through Change Control.
Original and Approved URS along with DQ provided by the Vendor shall be submitted to Quality Assurance Department.
DESIGN QUALIFICATION :
Design Qualification shall be prepared based on DQ / URS documents and information provided by the vendor.
Design Qualification shall be prepared as per format shown in Annexure-I.
Content of Header and Footer: refer point no. 5.2.24.
Click the link for download word file copy of this document:
https://pharmaguidehub.com/product/standard-operating-procedure-on-validation-and-qualification-procedure-for-facility-utility-equipment-and-process/
Content of Body Part:
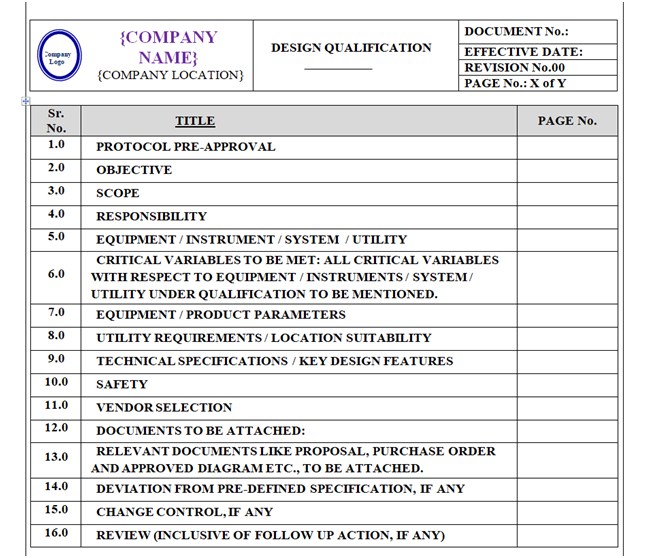
The format of Design Qualification shall contain the following sections but not limited to:
Equipment / Instrument / Utility Qualification:
- Pre-Approval
- Objective
- Scope
- Responsibility
- Equipment / Instrument / System / Utility
- Critical Variablesto be met:All critical variables with respect to Equipment / Instruments / System / Utility under qualification to be mentioned.
- Equipment / Product Parameters
- Utility Requirements / Location Suitability
- Technical Specifications / Key Design Features
- Safety
- Vendor Selection
- Documents to be attached:
- Relevant Documents like Proposal, Purchase Order and Approved Diagram etc., to be attached.
- Deviation from pre-defined specification, if any
- Change control, if any
- Review (inclusive of follow up action, if any)
- Conclusion
- Recommendation
- Abbreviations
- Revision history
Area Qualification:
- Protocol Pre-Approval
- Objective
- Scope
- Responsibility
- Project Requirement
- Design Qualification
- Documents to be attached
- Non compliance
- Change Control, if any
- Review (inclusive of follow up action, if any)
- Recommendation
- Conclusion
- Abbreviations
- Revision History
Water System:
- Protocol Pre-Approval
- Objective
- Scope
- Responsibility
- Project Requirements
- Brief Process Description
- User Requirement Specification
- Basis of System Design (For Purified Water Generation & Distribution System)
- Basic Treatment Scheme and System Description
- Basis of Design for Storage and Distribution of Purified Water
- Critical Variables to be Met
- Documents to be attached
- Deviation From Pre-Defined Specification, if any
- Change Control, if any
- Review (inclusive of follow up action, if any)
- Conclusion
- Recommendation
- Abbreviations
- Revision History
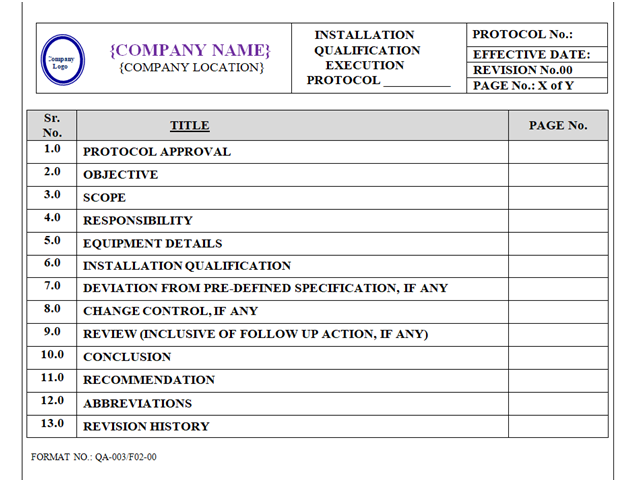
INSTALLATION QUALIFICATION PROTOCOL (IQ):
Installation Qualification Protocol shall be prepared as per format shown in Annexure-II.
Content of Header and Footer: These are mentioned in subsequent paragraphs
Content of Body Part:
Format of Installation Qualification Protocol shall contain the following information but not limited to:
Equipment / Instrument / Utility Qualification:
| Protocol approval |
| Objective |
| Scope |
| Responsibility |
| Equipment details |
| Installation qualification |
| Deviation from pre-defined specification if any |
| Change control, if any |
| Review (inclusive of follow up action, if any) |
| Conclusion |
| Recommendation Abbreviations Revision history |
Area Qualification:
- Protocol Pre-Approval
- Objective
- Scope
- Responsibility
- Project Requirement
- Design Qualification
- Documents to be attached
- Non compliance
- Change Control if any
- Review (inclusive of follow up action, if any)
- Recommendation
- Conclusion
- Abbreviations
- Revision History
Water System:
- Protocol Pre-Approval
- Objective
- Scope
- Responsibility
- Water System Details
- Purified Water System Description
- Critical Variables to be met
- References
- Documents to be attached
- Deviation from Pre-defined Specification if any
- Change Control, if any
- Review (inclusive of follow up action, if any)
- Conclusion
- Recommendation
- Abbreviations
- Protocol Post Approval
Revision History
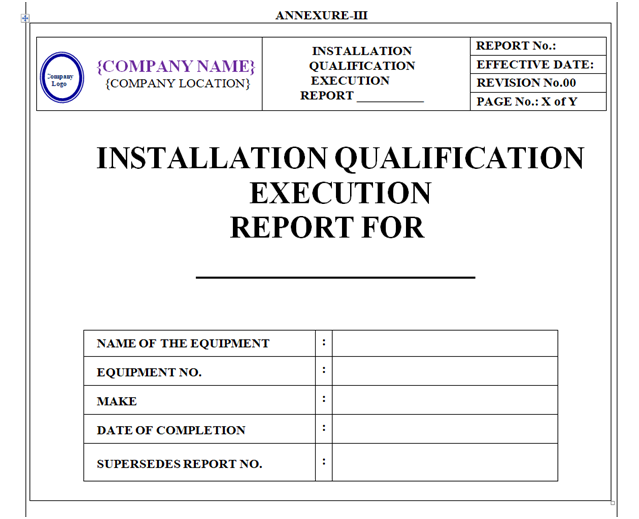
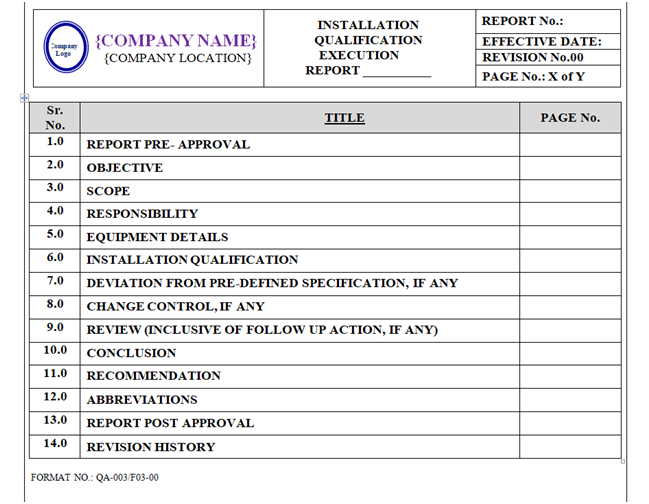
INSTALLATION QUALIFICATION REPORT (IQ):
Based on IQ protocol Qualification report shall be prepared as per format shown in Annexure-III.
Content of Header and Footer: These are mentioned in subsequent paragraphs
Content of Body Part:
Format of Installation Qualification Report shall contain the following information but not limited to:
Click the link for download word file copy of this document:
https://pharmaguidehub.com/product/standard-operating-procedure-on-validation-and-qualification-procedure-for-facility-utility-equipment-and-process/
Equipment / Instrument / Utility Qualification:
- Report Pre-Approval
- Objective
- Scope
- Responsibility
- Equipment Details
- Installation Qualification
- Deviation from Pre-defined Specification if any
- Change Control, if any
- Review (inclusive of follow up action, if any)
- Conclusion
- Recommendation
- Abbreviations
- Report Post Approval
- Revision History
Area Qualification:
- Report Pre-Approval
- Objective
- Scope
- Responsibility
- Project Requirement
- Design Qualification
- Documentstobeattached
- Non compliance
- Change Control, if any
- Review (inclusive of follow up action, if any)
- Recommendation
- Conclusion
- Abbreviations
- Report Post Approval
- Revision History
Water System:
- Report Pre-Approval
- Objective
- Scope
- Responsibility
- Water System Details
- Purified Water System Description
- Critical Variables to be met
- References
- Documents to be attached
- Deviation from Pre-defined Specification if any
- Change Control if any
- Review (inclusive of follow up action, if any)
- Conclusion
- Recommendation
- Abbreviations
- Report Post Approval
- Revision History
During and after installation of AHU duct the external area of the ductshall be check. So any gap between the ducts is not there and ensures external part should be affixed properly. Any extraneous part should not be exposed to outside the designated area of duct.
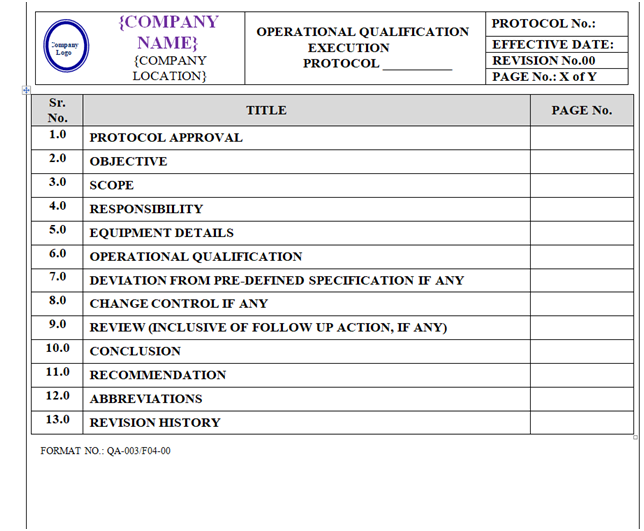
OPERATIONAL QUALIFICATION PROTOCOL:
Operational Qualification Protocol shall be prepared as per format shown in Annexure-IV.
Content of Header and Footer: are mention in above paragraphs.
Content of Body Part:
The format of Operational Qualification Protocol shall contain the following information but not limited to:
Equipment / Instrument / Utility Qualification:
| Protocol Approval |
| Objective |
| Scope |
| Responsibility |
| Equipment details |
| Operational qualification |
| Deviation from Pre-Defined Specification if any |
| Change control if any |
| Review (inclusive of follow up action, if any) |
| Conclusion |
| Recommendation |
| Abbreviations |
| Revision History |
Area Qualification:
- Protocol Pre-Approval
- Objective
- Scope
- Responsibility
- Operational Qualification
- Documents to be attached
- Non compliance
- Change Control, if any
- Review (inclusive of follow up action, if any)
- Recommendation
- Conclusion
- Abbreviations
- Revision History
Water System:
- Protocol Pre-Approval
- Objective
- Scope
- Responsibility
- Water System Details
- Critical Variable to be met
- References
- Documents to be attached
- Deviation from Pre-defined Specification, if any
- Change Control, if any
- Review (inclusive of follow up action, if any)
- Conclusion
- Recommendation
- Abbreviations
- Protocol Post Approval
OPERATIONAL QUALIFICATION REPORT:
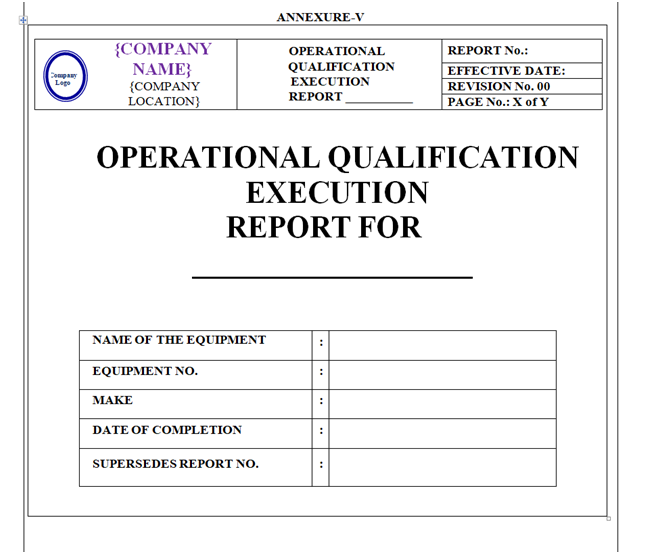
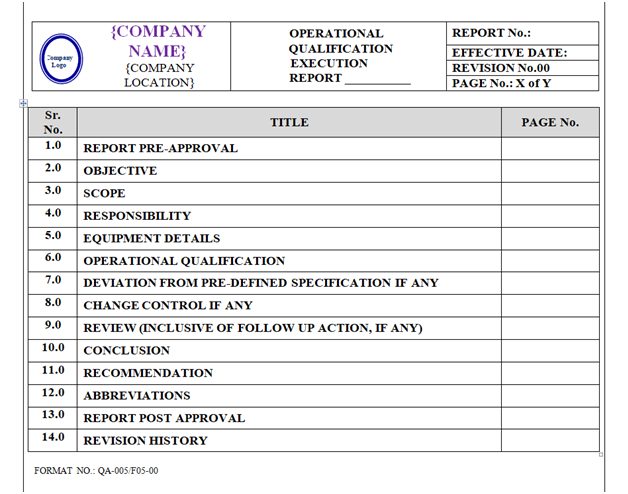
Based on Operational Qualification Protocol Qualification report shall be prepared as per format shown in Annexure-V.
Content of Header and Footer: are mention in above paragraphs.
Content of Body Part: The format of Operational Qualification Report shall contain the following information but not limited to:
Equipment / Instrument / Utility Qualification:
| Report Pre-Approval |
| Objective |
| Scope |
| Responsibility |
| Equipment details |
| Operational qualification |
| Deviation from Pre-defined Specification if any |
| Change control if any |
| Supporting attachments |
| Review (inclusive of follow up action, if any) |
| Conclusion |
| Recommendation |
| Abbreviations Report Post Approval |
| Revision History |
Area Qualification:
- Protocol Pre-Approval
- Objective
- Scope
- Responsibility
- Operational Qualification
- Documents to be attached
- Non compliance
- Change Control, if any
- Review (inclusive of follow up action, if any)
- Recommendation
- Conclusion
- Abbreviations
- Report Post Approval
- Revision History
Water System:
- Protocol Pre-Approval
- Objective
- Scope
- Responsibility
- Water System Details
- Critical Variable to be met
- References
- Documents to be attached
- Deviation from Pre-defined Specification, if any
- Change Control, if any
- Review (inclusive of follow up action, if any)
- Conclusion
- Recommendation
- Abbreviations
- Report Post Approval
- Revision History
On completion of Installation Qualification of system, Operational Qualification shall be started.
On review of IQ & OQ documents provided by Vendor, if any deficiency is observed, same shall be discussed with vendor and such activity shall either be completed by vendor or undertaken by {Company Name} {Company Location}, and documented.
PERFORMANCE QUALIFICATION (PQ):
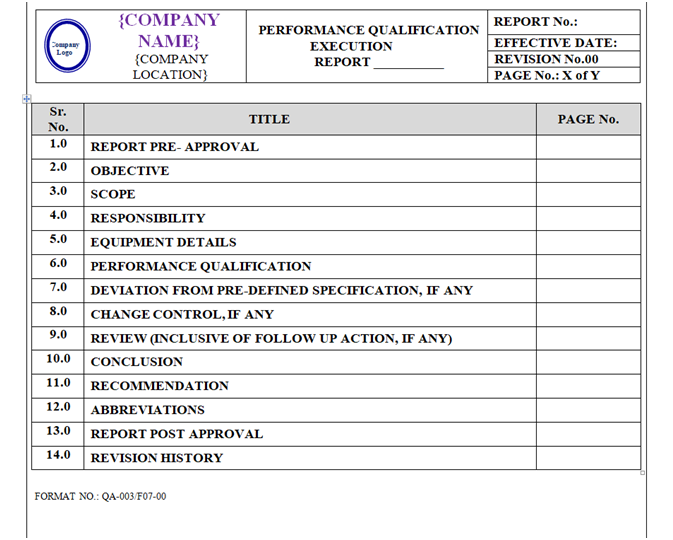
PERFORMANCE QUALIFICATION PROTOCOL:
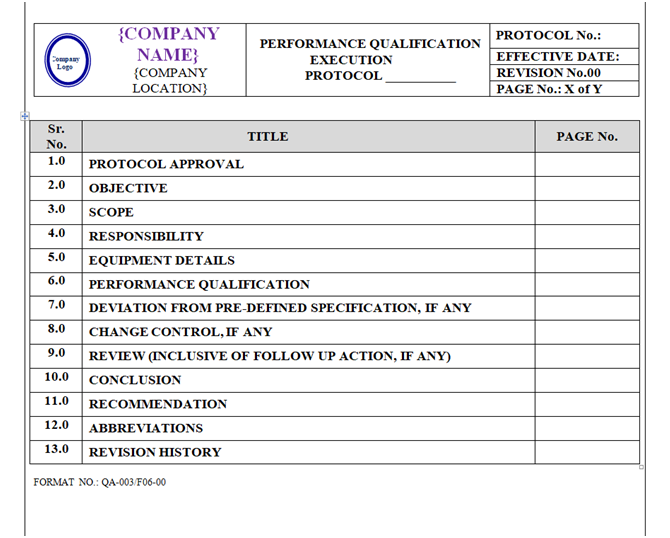
Performance Qualification Protocol shall be prepared as per format shown in Annexure-VI.
Content of Header and Footer: are mention in above paragraphs.
Content of Body Part:
The format of Performance Qualification Protocol shall contain the following information but not limited to:
Equipment / Instrument / Utility Qualification:
| Protocol approval |
| Objective |
| Scope |
| Responsibility |
| Equipment details |
| Performance qualification |
| Deviation from pre-defined specification if any |
| Change control if any |
| Review (inclusive of follow up action, if any) |
| Conclusion |
| Recommendation Abbreviations Revision history |
Water System Qualification:
- Protocol Approval
- Objective
- Scope
- Responsibility
- System Description
- Flow Diagram of Generation of Water
- Sampling Point Location and Specification for water validation Phase-I, II and III.
- Measurements
- Validation Procedure
- Validation & Sampling
- Validation Phase-I & II
- Sampling Plan for Water System Validation Phase I & II
- Validation Phase-III
- Sampling Plan for Water System Validation Phase-III
- Sampling Procedure & Testing
- Method of Sampling
- Sampling of Water for Chemical Analysis
- Sampling of Water for Microbiological Analysis
- Testing
- Action & Alert Limits
- Sanitization of Water System
- Qualification of Water System
- System Pre-requisites
- Acceptance Criteria
- Re-qualification Criteria and Change Control
- Ongoing System Monitoring
- References
- Documents to be Attached
- Non Compliance
- Deviation from Pre-defined Specification if any
- Change Control if any
- Annexure
- Abbreviations
- Revision History
Area Qualification:
- Protocol Approval
- Objective
- Scope
- Responsibility
- Project Requirement
- Performance Qualification
- Non Compliance
- Deviation from Pre-defined Specification, if any
- Change Control, if any
- References
- Documents to be attached
- Abbreviations
- Revision History
Click the link for download word file copy of this document:
https://pharmaguidehub.com/product/standard-operating-procedure-on-validation-and-qualification-procedure-for-facility-utility-equipment-and-process/
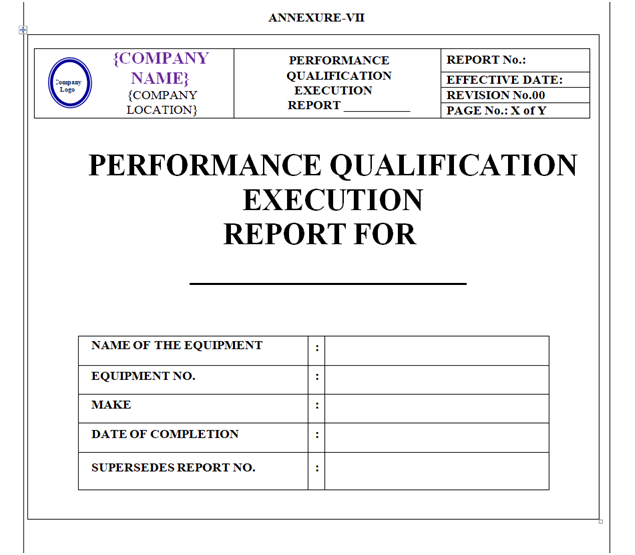
PERFORMANCE QUALIFICATION REPORT:
On the basis of Qualification Protocol, Qualification Report shall be prepared as per Annexure-VII.
Content of Header and Footer: are mention in above paragraphs.
Content of Body Part:
The format of Performance Qualification Report shall contain the following information but not limited to:
Equipment / Instrument / Utility Qualification Report:
| Pre-Approval |
| Objective |
| Scope |
| Responsibility |
| Equipment details |
| Performance qualification |
| Deviation and corrective action |
| Change control, if any |
| Review (inclusive of follow up action, if any) |
| Conclusion |
| Recommendation |
| Abbreviation |
| Post approval signature |
| Revision history |
Water System Qualification:
- Report Pre-Approval
- Objective
- Scope
- Responsibility
- Validation Phase-I & II
- Sampling Plan for Water System Validation Phase I & II
- Validation Phase-III
- Sampling Plan for Water System Validation Phase-III
- Sampling Procedure & Testing
- Method of Sampling
- Sampling of Water for Chemical Analysis
- Sampling of Water for Microbiological Analysis
- Testing
- Action & Alert Limits
- Sanitization of Water System
- Qualification of Water System
- System Pre-requisites
- Acceptance Criteria
- Re-qualification Criteria and Change Control
- Ongoing System Monitoring
- References
- Documents Attached
- Non Compliance
- Deviation from Pre-defined Specification, if any
- Change Control, if any
- Annexure
- Review (inclusive of follow up action, if any)
- Conclusion
- Recommendation
- Abbreviations
- Report Post Approval
- Revision History
Area Qualification:
- Report Pre-Approval
- Objective
- Scope
- Responsibility
- Performance Qualification
- Non Compliance
- Deviation from Pre-defined Specification, if any
- Change Control, if any
- References
- Documents attached
- Review (inclusive of follow up action, if any)
- Conclusion
- Recommendation
- Abbreviations
- Report Post Approval
The Qualification Report shall be reviewed by Head of User Department, QC / Engineering / Production Head (as applicable) and approved by Head of QA. The approval of Qualification Report is considered as handover of the system to user Department for its intended purpose.
NUMBERING SYSTEM OF VALIDATION PROTOCOL AND REPORTS:
The Numbering system of validation protocol and reports shall be assigned as:
The validation Protocol No. shall be assigned as:
XXX/YYY
Where,
XXX: Indicate for name of protocol e.g. (Process validation protocol (PVP), Hold time study protocol (HTSP), Analytical method validation protocol (AMVP), Cleaning validation protocol (CVP) Stability study protocol (SSP)
YYY: Indicate serial No. start from 001, 002, 003, 004…………………
For Process Validation Protocol: E. g. PVP/001,
For Hold time study Protocol: E. g. HTSP/001
For analytical method validation Protocol: E.g. AMV/001
For cleaning validation Protocol: CVP/001 For stability study Protocol: SSP/001
The validation Report No. shall be assigned as:
XXX/YYY
Where,
XXX: Indicate for name of protocol e.g. (Process validation Report (PVR), Hold time study Report (HTSR), Analytical method validation Report (AMVR), cleaning validation Report (CVR) Stability study Report (SSP)
YYY: Indicate serial No. start from 001, 002, 003, 004…………………
For Process Validation Report: E. g. PVR/001
For Hold time study Report: E. g. HTSR/001
For analytical method validation Report: E.g. AMVR/001
For cleaning validation Report: CVR/001
For stability study Report: SSR/001
The numbering system for revalidation protocol and report
XXXX/YYY
XXXX: Indicate re-validation protocol name.
YYY: Indicate serial No. start from 001, 002, 003, 004……………….
E.g. RPVP/001
PREPARATION OF VALIDATION / RE-VALIDATION PROTOCOLS:
Process validation Protocols and reports shall be prepared as per relevant SOP. The format of process validation Protocol and report shall contain the following information.
PROCESS VALIDATION PROTOCOL:
Process Validation Protocol shall contain the following Heads but not limited to:
- Protocol approval
- Objective
- Scope
- Responsibility
- Reason for validation
- Process validation methodology
- Training
- Master document verification
- Bill of material
- Process validation pre-requisite
- Product information
- Specifications and standard test procedure
- Equipment qualification verification
- Environment condition of area
- Process flow diagram
- Process steps and critical process variables for validation
- Methodology and sampling procedure for each stage
- Critical quality attribute (CQA) and analysis plan
- Process validation acceptance criteria
- Revalidation
- Deviation
- Validation stability plan
- Validation report
- Conclusion
- References
- List of attachments
- Abbreviations
- Revision history
PROCESS VALIDATION REPORT:
| Report pre- approval |
| Objective |
| Scope |
| Responsibility |
| Reason for validation |
| Training record |
| Document verification |
| Raw materials & packaging material dispensing verification: |
| Critical process variables |
| Temperature, relative humidity, pressure differential record during curing |
| Temperature, relative humidity, pressure differential record during inspection |
| Test result |
| Yield |
| Non conformance |
| Deviation from predefined specification |
| Change control, if any |
| Review (inclusive of follow up action |
| Conclusion |
| Recommendation |
| Abbreviation |
| Report post approval |
| Revision history |
Hold Time Study Protocol (For Product):
Hold Time Study Protocol and report format shall be prepared as per relevant SOP. The hold time study protocol and repot shall contain the following contents:
| Protocol approval |
| Introduction |
| · Objective |
| Scope |
| Responsibility |
| Training record |
| Detail of semi-finished (naked and pouched gummies) and selection criteria |
| Procedure |
| Sampling plan |
| Acceptance criteria for dispensed raw material |
| Acceptance criteria for semi-finished (naked gummies and pouched gummies) |
| Analytical method |
| Protocol certification |
| Approval of results |
| Revalidation criteria |
| References |
| Documents to be attached |
| Non conformance |
| Deviation from pre-defined specification if any |
| Change control if any |
| Review (inclusive of follow up action, if any) |
| Conclusion |
| Recommendation |
| Abbreviations |
| Revision history |
Hold Time Study Report (For Product):
| Report pre- approval |
| Introduction |
| Objective |
| Scope |
| Responsibility |
| Reason for qualification |
| Pre-qualification requirement |
| Qualification test result |
| Non conformance |
| Deviation from predefined specification |
| Change control if any |
| Review (inclusive of follow up action) |
| Conclusion |
| Recommendation |
| Abbreviation |
| Report post approval |
| Revision history |
Analytical Method Validation Protocol:
Analytical Method Validation Protocol and report shall be prepared as per relevant SOP. The AMV Protocol and report shall contain the following contents:
- Validation Team
- Objective
- Scope
- Purpose
- Responsibility
- Training Details
- Product Details
- Selection for analytical performance parameter for AMV
- Definition
- Procedure for method validation
- Validation Acceptance Criteria
- References
- Summary Report
- Attachments
- Deviation, if any
- Conclusion
- Recommendation
- Abbreviations
- Revision History
Analytical Method Report
- Validation Team
- Objective
- Scope
- Purpose
- Responsibility
- Training Details
- Product Details
- Selection for analytical performance parameter for AMV
- Definition
- Procedure for method validation
- Validation Acceptance Criteria
- References
- Summary Report
- Attachments
- Deviation, if any
- Conclusion
- Recommendation
- Abbreviations
- Report Post Approval
- Revision History
Stability Study Protocol:
The Stability study Protocol and report format shall be prepared as per relevant SOP. The stability study protocol and report shall contain following contents as given below:
| Protocol approval |
| Objective |
| Scope |
| Responsibility |
| Training record of concerned personnel |
| Product information |
| Sample quantity for testing |
| Reason for stability study |
| Specification and test methodology information |
| Types of studies |
| Test results acceptance criteria Specification |
| Reference to analytical validation (stability indicating) |
| Sample charging and sample plan |
| Stability study report |
| Out of specification |
| Deviations / incidents |
| Change control |
| Equipment and storage malfunction |
| Commitment |
| Conclusion |
| Reference documents |
| Abbreviations |
| Revision History |
Stability Study Report:
Stability Study Report shall contain the following heads but not limited to:
| Report pre-approval |
| Objective |
| Scope |
| Responsibility |
| Training record of concerned personnel |
| Product information |
| Accelerated stability study test result |
| Long term stability study test result |
| Non conformance |
| Deviation from predefined specification |
| Change control, if any |
| Review (inclusive of follow up action) |
| Conclusion |
| Recommendation |
| Abbreviations |
| Report post approval |
| Revision History |
Cleaning Validation Protocol:
Cleaning Validation Protocol and report format shall be prepared as per relevant SOP. The cleaning validation protocol and report shall contain the following contents as given below:
| Protocol approval |
| Objective |
| Scope |
| Responsibility |
| Training details |
| Validation approach for campaign production, cleaning validation & hold time study |
| Criteria for selection of product for campaign production |
| Elements of cleaning process validation |
| Methodology of cleaning process validation |
| Acceptance criteria |
| Cleaned equipment hold time study |
| Environment monitoring |
| Testing method reference |
| Acceptance criteria |
| Re-validation criteria |
| References |
| Documents to be attached |
| Non conformance |
| Deviation from pre-defined specification, if any |
| Change control if any |
| Review (inclusive of follow up action, if any) |
| Conclusion |
| Recommendation |
| Abbreviations |
| Revision history |
Cleaning Validation Report
| Report pre-approval |
| Objective |
| Scope |
| Responsibility |
| Reason for validation |
| Pre-validation requirement |
| Documents verification |
| Cleaning validation study |
| Documents attached |
| Non conformance |
| Deviation from pre-defined specification, if any |
| Change control, if any |
| Review (inclusive of follow up action, if any) |
| Conclusion |
| Recommendation |
| Abbreviations |
| Report post approval |
| Revision history |
System / Utility / other Validation Protocol:
System / Utility / Thermal Mapping / BET Validation and any Others Validation Protocol shall contain the following Heads but not limited to:
- Protocol Approval
- Objective
- Scope
- Responsibility
- Reason for Validation
- Site of Study
- Training Details
- Methodology
- Master Document Verification
- Acceptance Criteria
- Validation / Re-Validation Criteria
- Frequency of Validation
- Change Control and Deviation
- Conclusion
- Recommendation
- References
- Documents to be Attached
- Deviation From Pre-Defined Specification, If any
- Change Control, If any
- Abbreviations
- Revision History
System / Utility / other Validation Report:
System / Utility / Thermal Mapping and any Others ValidationReport shall contain the following heads but not limited to:
- Report Pre Approval
- Objective
- Scope
- Responsibility
- Reason for Validation
- Site of Study
- Training Details
- Methodology
- Master Document Verification
- Acceptance Criteria
- Validation / Re-Validation Criteria
- Frequency of Validation
- Change Control and Deviation
- Conclusion
- Recommendation
- References
- Documents to be attached.
- Deviation From Pre-Defined Specification If any
- Change Control If any
- Abbreviations
- Report Post Approval
- Revision History
LAYOUT OF PROTOCOL CUM REPORTS, PROTOCOLS & REPORTS:
All Protocols & Reports shall contain Header, Footer and Body
The Line Spacing between two points or title & subtitle shall be 1.0 / 1.5 and font style shall be Times New Roman 12 font.
Paper Width 8.5”, Height 11.5” (Paper Size Custom) & Margin Top, Left, Right 24pt & Bottom Margin 31pt.
Protocols & Reports (Master Copy) shall be printed on A4 size Off-white colored Executive Bond Paper using “Times New Roman” Font with colour Ink.
Printing shall be done on one side of the paper only.
All Protocols & Reports contents shall be covered by Single Borderline (Line width ½ pt).
FONT SIZE OF HEADER, FOOTER & BODY CONTENTS:
| NAME OF CONTENT | FONT SIZE |
| HEADER: | |
| Name of the Organization | 16 Bold & Capital |
| Location (Company Location) | 12 Normal & Capital |
| Logo with {Company Name} (On Left Hand Side Corner of The Page) | Logo– Height-0.75’’& Width-0.66’’ |
| Title of URS, Protocol & Report | 12 Bold & Capital |
| URS / Protocol / Report No. | 12 Bold & Capital |
| Effective Date | 12 Bold & Capital |
| Page No. (In the form of X of Y) | 12 Bold & Capital |
| FOOTER: | |
| Format No. | 09 Capital Normal |
| BODY: | |
| Main Heading & Table Heading | 12 Bold & Capital |
| Sub Heading | 12 Normal & Bold |
| Table Contents Except Headings | 12 Normal |
| FIRST PAGE CONTENT: | |
| Title of Protocol & Report | 28 Bold & Capital with Double Line Space |
| Table Content | 12 Bold & Capital |
STORAGE OF PROTOCOLS & REPORTS:
Master Qualification Protocols and Reports shall be stored in Quality Assurance Department.
If Protocol is revised, previous version of Protocol shall be made Obsolete and shall be archived for Historical purpose as per Document Retention Policy.
CONTROL, DISTRIBUTION & EXECUTION OF PROTOCOLS & REPORTS:
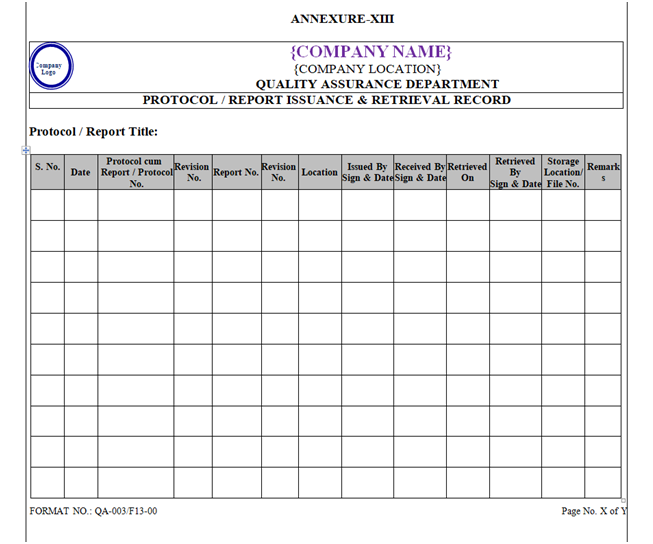
Controlled Copy of Protocols & Reports for Execution shall be distributed to respective Department by QA and details shall be recorded in format “Protocol / Report Issuance & Retrieval Record” as shown in Annexure-XIV.
QA shall put the ‘Controlled Copy Stamp” on each page of Protocols & Reports& distributed to different Departments for Execution Purpose.
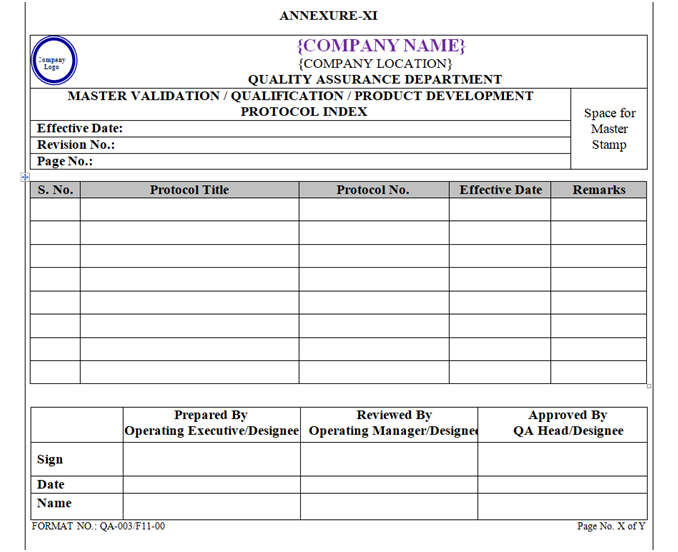
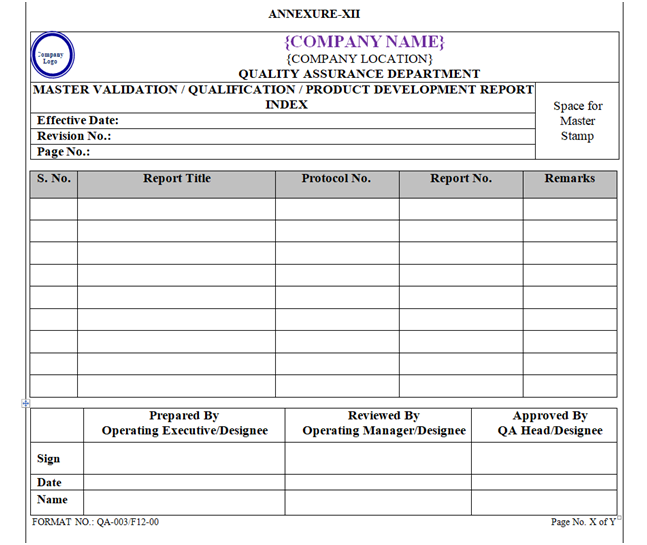
A Master Index of Protocols & Reports shall be maintained by QA Department in format “Master Validation / Qualification Report Index” as shown in Annexure-XII, XIII.
The Index of Protocols and Reports shall be updated at the frequency of two Year or whenever required, manual corrections shall not be allowed in this Index.
In case of discontinuation of the Protocol and Report, the Protocol and Report may be discontinued; however the same Protocol and Report No. shall not be allotted to any newly prepared Protocol and Report.
All Master Copy of Protocols and Reports shall be stamped as “MASTER COPY” in red ink on all the pages. Specimen of Master Copy stamp is shown in SOP No.: QA/001.
The Execution Copy of Protocol shall be distributed by making Photostat copies of the Master Copy and shall be stamped as “CONTROLLED COPY” in green ink at the Right Upper Corner on all pages of Protocol. Specimen of Controlled Copy stamp is shown in SOP No.: QA/001.
In case, if any Protocol and Report is submitted to the External Agencies (i.e. Regulatory, Customers etc.) a photocopy of Master Copy / Executed Copy shall be made by QA and shall be stamped as “UNCONTROLLED COPY” in Violet ink on Left Corner of the page below footer.
The Copy of Protocol and Report that is meant only for Information / Reference purpose shall be made by Photostat of Master Copy / Executed Copy (in case of executed Protocol / Report).
If Soft Copy of Protocol and Report is forwarded, it shall be submitted as “Read only” protected PDF format.
Protocols & Reports shall be revised only if any change is required, if there is no change continue with same protocol & report.
Revision of Protocol and Report shall be regulated through “Change Control Procedure”. The Obsolete Copy shall be stamped as “OBSOLETE COPY” in Black Ink in the Left Upper Corner of all the pages. Specimen of Obsolete Copy Stamp is shown in SOP No.: QA/001.
The Master Copy of Protocol and Report which is discontinued, or which is cancelled due to implementation of any new Protocol and Report shall be stamped as “CANCELLED COPY” in Black Ink at the Left Upper Corner of all the pages.
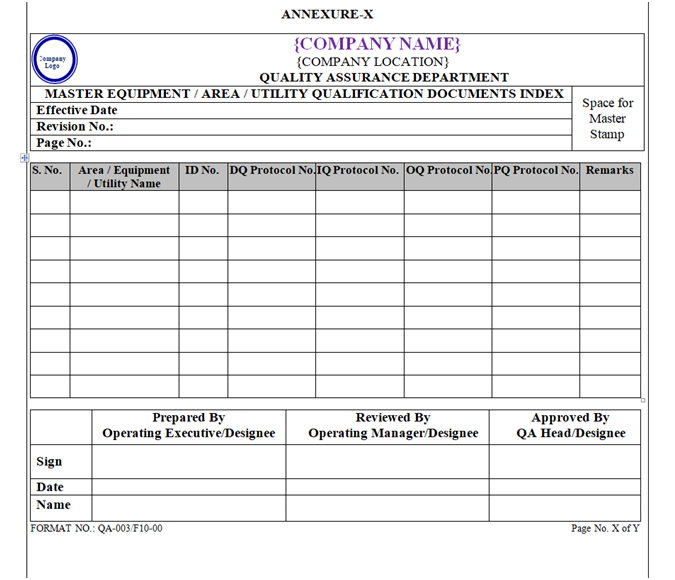
Index of Qualification Documents shall be prepared as per Format shown in Annexure-, X, XI, XII, and XIII.
DESTRUCTION OF PROTOCOLS & REPORTS:
All Qualification & Validation Documents (i.e. Process Validation Protocols / Reports, Stability Data & Documents, Qualification & Validation Documents for Critical Utilities / System / Equipments etc.) shall be retained till the life of the Organization.
REFERENCES:
A WHO guide to Good Manufacturing Practice (GMP) requirements (Part 2: Validation).
QA SOP No. QA/001, “Preparation, Approval, Control, Distribution, Revision, Retrieval & Destruction of Standard Operating Procedure”.
Click the link for download word file copy of this document:
https://pharmaguidehub.com/product/standard-operating-procedure-on-validation-and-qualification-procedure-for-facility-utility-equipment-and-process/
ANNEXURES:
| ANNEXURE No. | TITLE OF ANNEXURE | FORMAT No. |
| Annexure-I | Protocol For Design Qualification | QA-003/F01-00 |
| Annexure-II | Protocol For Installation Qualification | QA-003/F02-00 |
| Annexure-III | Report For Installation Qualification | QA-003/F03-00 |
| Annexure-IV | Protocol For Operational Qualification | QA-003/F04-00 |
| Annexure-V | Report For Operational Qualification | QA-003/F05-00 |
| Annexure-VI | Performance Qualification Protocol | QA-003/F06-00 |
| Annexure-VII | Performance Qualification Report | QA-003/F07-00 |
| Annexure-VIII | User Requirement Specification | QA-003/F08-00 |
| Annexure- IX | User Requirement Specification Index | QA-003/F09-00 |
| Annexure-X | Master Equipment / Area / Utility Qualification Documents Index | QA-003/F10-00 |
| Annexure-XI | Master Validation / Qualification Protocol Index | QA-003/F11-00 |
| Annexure-XII | Master Validation / Qualification Report Index | QA-003/F12-00 |
| Annexure-XIII | Protocol / Report Issuance & Retrieval Record | QA-003/F13-00 |
DISTRIBUTION:
- Controlled Copy No. 01 : Head Quality Assurance
- Controlled Copy No. 02 : Head Quality Control
- Controlled Copy No. 03 : Head Production
- Controlled Copy No. 04 : Head Engineering
- Controlled Copy No. 05 : Head Warehouse
- Master Copy : Quality Assurance Department
ABBREVIATIONS :
| Ltd. | : | Limited |
| SOP | : | Standard Operating Procedure |
| No. | : | Number |
| QA | : | Quality Assurance |
| QC | : | Quality Control |
| S. No. | : | Serial Number |
| HPLC | : | High Performance Liquid Chromatography |
| FS | : | Functional Specification |
| DS | : | Design Specification |
| VMP | : | Validation Master Plan |
| IT | : | Information Technology |
| API | : | Active Pharmaceutical Ingredients |
| SS | : | Stainless Steel |
| AHU | : | Air Handling Unit |
| GPT | : | Growth Promotion Test |
| : | Portable Document Format | |
| ID No. | : | Identification Number |
| URS | : | User Requirement Specification |
| DQ | : | Design Qualification |
| IQ | : | Installation Qualification |
| OQ | : | Operational Qualification |
| PQ | : | Performance Qualification |
REVISION HISTORY:
CHANGE HISTORY LOG
| Revision No. | Details of Changes | Reason for Change | Effective Date |
| 00 | New SOP | Not Applicable | To Be recorded Manual. |
ANNEXURE-I

ANNEXURE – II

ANNEXURE – III
ANNEXURE – IV

ANNEXURE – V
ANNEXURE – VI

ANNEXURE – VII
ANNEXURE – VIII


ANNEXURE – IX
ANNEXURE – X
ANNEXURE – XI
ANNEXURE – XII
ANNEXURE – XIII
Click the link for download word file copy of this document:
https://pharmaguidehub.com/product/standard-operating-procedure-on-validation-and-qualification-procedure-for-facility-utility-equipment-and-process/



Thanks for your help and for writing this post.
Your articles are very helpful to me. May I request more information?
Thank you for providing me with these article examples.
Thank you for your post. I really enjoyed reading it, especially because it addressed my issue. It helped me a lot and I hope it will also help others.
Your articles are extremely helpful to me. Please provide more information!
Thank you for your help and this post. It’s been great.
Thank you for your post. I really enjoyed reading it, especially because it addressed my issue. It helped me a lot and I hope it will also help others.
You’ve been great to me. Thank you!
Can you write more about it? Your articles are always helpful to me. Thank you!
Please tell me more about this. May I ask you a question?
Thank you for writing this article. I appreciate the subject too.
Attractive section of content. I just stumbled upon your
site and in accession capital to assert that I get in fact
enjoyed account your blog posts. Any way I’ll be
subscribing to your augment and even I achievement you access consistently quickly.
Hello, i think that i saw you visited my site so i came to
“return the favor”.I am attempting to find things to improve my web site!I suppose its ok to
use some of your ideas!!
You’ve been great to me. Thank you!
You helped me a lot by posting this article and I love what I’m learning.
Hi, just wanted to mention, I enjoyed this blog
post. It was inspiring. Keep on posting!
Thank you for sharing this article with me. It helped me a lot and I love it.
Excellent post. I used to be checking continuously this blog and I am impressed!
Very helpful information specially the closing section 🙂 I care for such information a lot.
I was seeking this particular information for a very long time.
Thank you and best of luck.
Hi there to every single one, it’s in fact a pleasant for me
to pay a visit this web page, it consists of helpful Information.
I used to be recommended this web site by my
cousin. I’m no longer sure whether this submit is written through him ass nobody else understand such
certain about my trouble. You are amazing! Thanks!
No matter if some one searches for his essential thing, so he/she needs
to be available that in detail, so that thing is maintained over here.
Thanks for posting. I really enjoyed reading it, especially because it addressed my problem. It helped me a lot and I hope it will help others too.
Thank you for your articles. They are very helpful to me. Can you help me with something?
Thanks for posting. I really enjoyed reading it, especially because it addressed my problem. It helped me a lot and I hope it will help others too.
That is a really good tip particularly to those fresh to
the blogosphere. Simple but very precise information… Appreciate your sharing this one.
A must read article!
May I request that you elaborate on that?
Your posts have been extremely helpful to me. Thank you!
The articles you write help me a lot and I like the topic
I was suggested this blog by way of my cousin. I’m no longer certain whether this
putt up is written via him as nobody else recognise such designated approximately my difficulty.
You are wonderful! Thanks!
Can you tell us more about this? I’d care to find out
more details.
Thank you for sharing this article with me. It helped me a lot and I love it.
Please provide me with more details on the topic
Howdy I am so thrilled I found your blog page,
I really found you by accident, while I was searching on Google for something else, Regardless I am here now and would just
like to say kudos for a tremendous post and a all round enjoyable blog (I also love the theme/design), I don’t
have time to go through it all at the moment but I have bookmarked it and also included
your RSS feeds, so when I have time I will be back to read a great deal more, Please do keep up the excellent job.
If some one needs expert view concerning blogging after that i advise him/her to visit this webpage, Keep
up the good job.
Hi! I’ve been following your website for a long time now and finally got the courage
to go ahead and give you a shout out from Atascocita Tx!
Just wanted to mention keep up the fantastic job!
You helped me a lot by posting this article and I love what I’m learning.
Thanks for posting. I really enjoyed reading it, especially because it addressed my problem. http://www.kayswell.com It helped me a lot and I hope it will help others too.
Awesome! Its actually remarkable article, I have got much clear idea regarding from this
paragraph.
Right now it seems like Drupal is the best blogging platform available right now. (from what I’ve read) Is that what you are using on your blog?
What’s up, all is going nicely here and ofcourse every one is sharing facts, that’s truly good,
keep up writing.
Somebody essentially assist to make critically posts I would state. That is the first time I frequented your website page and to this point? I amazed with the research you made to make this actual put up amazing. Magnificent task!