

- OBJECTIVE:
To lay down a standard operating procedure for the handling of deviations and incidents.
- SCOPE:
This SOP is applicable to all the deviations and incidents observed in Warehouse, Production, Engineering, Quality Control, Information Technology and Quality Assurance departments at {Company Name}, {Company Location}.
- RESPONSIBILITY:
- Initiation of Deviation and Incident Report:
The Deviation and Incident report may be initiated by following functions based on their observations related to:
- Quality Control: Manager/analyst/designee for sampling error, analytical issue, Observation in stability test, instrument calibration, qualification, maintenance issue, standardization, storage of WS/Reference standard, malfunctioning of any lab instrument (such as HPLC, UV, pH meter, stability chamber, balances etc.), analyst training issue, environmental conditions, safety, documentation, specifications, test procedures, art works, GLP compliance, analytical work sheet of raw, packing, in-process, intermediates and finished products, numbering systems, item code or any other observations related to laboratory activity.
- Production: Manager/executive/designee for dispensing error, labeling error, quantity verification, material identification, observation during manufacturing, process parameters, instrument calibration, qualification/maintenance issue, storage of in-process material, malfunctioning of any manufacturing equipment/instrument (including, but not limited to sifter, Mixing Tank, Manufacturing, curing, polishing m/c, coating pan, packing m/c, batch coding, packing balances, utility systems), equipment cleaning, environmental systems, personnel practices, training of the operating personnel, housekeeping, safety, documentation, GMP compliance, planning, batch scheduling, batch release or any other observations related to manufacturing or unit operation.
Warehouse: Manager/executive/designee for observations related to item code, observation on instrument calibration, qualification, maintenance issue, standardization of weights, storage conditions of material, malfunctioning of any equipment in warehouse, mix up, environmental conditions, safety,FIFO/FEFO system, documentation, GMP compliance, or any other observations related to warehouse activity.
Engineering: Manager/executive/designee for observation like not attaining manufacturing process parameters, instrument calibration, qualification, validation, preventive maintenance, break down, malfunctioning of any manufacturing equipment/instrument, utility systems, environmental conditions, personnel practices, training of the operating personnel, housekeeping post maintenance, safety, documentation, spare parts or any other observations related to any operations of utility systems and documentation.
Quality Assurance: Manager/executive/designee for review of validation protocol, site master file, master plans, instrument calibration, qualification, validations, training and audit calendars, vendor audits, external audit observation, QC/GLP systems, analytical data review, review of documents related to manufacturing, warehouse, engineering, personnel and administration, training records, technical agreements, documents submitted to Regulatory Affairs, batch documentation, conditional / quarantine release, art works, vendor qualification, requalification, dimensions, registration number and inventory issues, GMP compliance, delay in closures of investigation, verification of CAPA implementation and non conformance in any of the function which may have impact on assurance of product safety, efficacy, quality.
IT: Manager/executive/designee for software issues, SOPs related to IT, back up data archival documentation, qualifications.
Verification and investigation of the Deviation and Incident:
Manager / designee of initiating department to review and initiate investigation on the Deviation and Incident as per the procedure detailed out in the SOP and the information required to be furnished in the format and supporting information/data as applicable.
To review the investigational findings, review similar historical event, arrive to the cause or probable cause of the Deviation and Incident. If required, conduct the analysis to find the root cause which has resulted in to the particular situation.
Propose corrective and preventive actions.
Respective department head to evaluate the risk involved through impact assessment on the product quality and access the suitability of the corrective/preventive actions proposed by the investigating team/investigator.
Quality Assurance:
To help in formulating the investigation plan, participate in the investigation; evaluate the investigation findings and the corrective / preventive actions proposed. Evaluate the risk and impact. Finally recommend for authorization or rejection of the Deviation and Incident.
Quality assurance head to authorize or reject the Deviation and Incident based on the risk impact assessment and final disposition of the product/batch in question, if applicable.
To authorize for extension of the investigation.
To take support from R&D, supplier / vendor in investigation to arrive at the cause.
Closure of the reported deviation / incident with justification.
To log the deviation and incident.
QA shall be responsible to report the Deviation and Incident to senior management to seek their support depending on the requirement.
Compliances:
Concerned department head and QA head shall be responsible for implementation of the corrective and prevention action plan.
A to track the nature of deviations/Incident and to review the historical data for impact/risk assessment and advice for effectiveness of the CAPA.
Click the link for download word file copy of this document: https://pharmaguidehub.com/product/standard-operating-procedure-for-the-handling-of-deviation-and-incident/
After evaluation final decision on the Deviation and Incident shall be taken by QA head or his designee.
ACCOUNTABILITY:
QA Head shall be accountable for implementation of SOP.
- PROCEDURE:
- A deviation is an activity which is outside the written down procedure of an operation / activity, but may be implemented for completion of that activity/operation after due impact assessment and authorization by QA.
- An incident is an unexpected event that has occurred during the ongoing operation/activity documentation/entries at any stage of the receipt, storage, manufacturing, packaging, analysis and distribution of a drug product / intermediate / raw material / packaging material / drug substance.
This shall also include rejection of incoming raw material, packing material, abnormal on line rejections, yield variations, inadequate cleaning during batch manufacturing and packaging. These incidents are to be reported as and when they occur and to be investigated for impact assessment
- All the deviations / incidents shall be reported as and when observed
- The person who observes the deviations / incidents shall inform the concerned department head and quality assurance about the deviations / incidents for further action. The observer may be any person and he can raise the deviations / incidents report and immediate action and CAPA shall be given by concerned / respective department head.
- Any deviations / incidents observed shall be reported on deviations / incidents report as per Annexure-II.
- Issuance of deviations / incidents report and numbering system:
- The initiator shall initiate request to QA for issuance of deviations / incidents report.
- QA officer shall make copy of deviations / incidents report as per Annexure-II and put the sign with date on ‘Issued by’ on first pages & issue to initiator as per requirement.
- Every Deviation and Incident Report shall be allotted a number consisting of seven characters:
- DRYYXXX
- IRYYXXX
- Where, DR – Deviation Report
- IR – Incident Report
- YY – the current year 23….99, such as 23 for 2023
- XXX – Numerical number 001…..999, such as 001 for first deviation of the year
- Example: The first deviation occurring in the year 2023 shall be numbered as DR23001
- The first Incident occurring in the year 2023 shall be numbered as IR23001
- Deviation and Incident report shall be filled by the concerned functional head / designee with brief description of the nature of Deviation and Incident.
- A common deviation / incidences may be filled for similar nature of deviation / incident, if observed during review on a later date. Incident for discrepancy observed during documentation review or system review shall also be filled and assessed for its impact on the quality of the product.
- In case of deviation / incident related to incoming raw material / packaging materials, the investigation may be extended to the vendor’s site in order to find out the cause for the deviation/ incidents. This may be done through discussions with the vendor, process reviews, transportation methods or site audits. Accordingly CAPA shall be instituted at vendors end.
- Functional head along with the concerned officer shall investigate the reasons for the occurrence of the deviation/ incident and describe the findings of investigation and record the conclusions in the report.
- For investigation of deviation / incident, people from relevant sections / areas may be interviewed and/or counselled and the details of this interview / counselling shall be recorded in the Interview cum counselling form (if required) as per Annexure-V.
- The department head along with representative from QA will determine whether these deviations / incident are having any impact on the quality of the product.
- The functional & department head shall recommend the corrective and preventive actions to prevent reoccurrences of the same.
- The investigation report in the format attached shall be submitted to the quality assurance head. Quality assurance head shall in turn to verify the extent of impact and suitability of corrective and preventive actions.
- On the basis of this evaluation, the quality assurance head shall decide on authorization/ rejection on deviations/Incidents.
- It shall be the responsibility of QA to verify the compliance of corrective and preventive actions.
- The impact of the deviation and incident shall be evaluated on the other batches manufactured in the same campaign / equipment / area or using same raw materials / packaging material depending upon the investigation findings.
- Deviation / incident report shall be maintained by QA officer/executive with entries of each Deviation / incident reported as per Annexure-I. The Deviation / incident record shall be maintained yearly.
- A trend of Incidents, Deviations, Market complaints, OOS observations shall be maintained and this historical data shall be reviewed to identify the reoccurring instances and for drawing correlation and impact assessment. Such trends shall also be utilized to track the effectiveness of corrective and preventive actions taken time to time.
- The information compiled shall be reviewed and summary shall be referred in the Annual product quality review for related issues.
- All the investigations shall be completed within 30 days from the date of observation of the deviation / incident.
- In case the investigation is not completed within the given time, the same can be extended by QA head with justification as per Annexure-IV. If necessary support from R&D, vendor may be requested which may help in investigation and finding the cause.
Click the link for download word file copy of this document: https://pharmaguidehub.com/product/standard-operating-procedure-for-the-handling-of-deviation-and-incident/
- TRENDING OF DEVIATION AND INCIDENT:
- Tracking and trending of deviations/incident shall be carried out by QA.
- Trend shall be done based on the following criteria:
- Review of Incident and deviation based on their classification.
- Review of incident and deviation based on their closure status.
- Review of incident and deviation based on their quality Impact / Not Impact.
- Review of incident and deviation based on CAPA.
- Department wise review of Incident and Deviation.
- Review of Incident and Deviation based on root cause.
- Such trend shall be prepared & reviewed by QA on every six months. If less than three deviation and incident will happened in six months then trend shall be prepared & reviewed by QA on annually basis and suitable actions shall be taken as per Annexure-III.
- CATEGORY FOR DEVIATION AND INCIDENT:
- Critical: The deviation and incidents which has direct impact on product quality andstability.Areas such as contamination of APIs (during dispensing, charging, solution preparation) critical process parameters, equipment / instrument performance breakdown, utility failure, calibration of critical instruments, equipment cleaning, line clearance, cross contamination etc.
- Major: The deviation /incidents which has no direct impact on product quality and stability. Non critical process parameters, momentary variations in environmental condition, weighment of Excipients, calibration of non critical instruments, cleaning of components not coming in direct contact with product, batch coding, secondary packing etc.
- Minor: The deviation / incidents which has no impact on product quality /stability. Such as cleaning of non core areas, entry in documents / log books, over writing, issues related to tertiary packing, shortages, typographical errors, transcription errors, and chromatographic error.
- Deviation and Incident report is a specific one time use document and the original copy should be retained with quality assurance.
- All data related to the Deviation and Incident shall be attached to the Deviation and Incident report.
- The root cause of Deviation and Incident shall be investigated using SQC tools (cause of effect analysis or failure mode & effect analysis).
- The root cause(s) or the most probable cause(s) shall be identified by the investigation team, comprising of affected department expert, QA expert and other department support experts (as required).
- This can be through brain storming session and SQC tools as described above.
- Upon investigation / identification of the root cause(s), the investigation team shall identify corrective action(s) and preventive action(s) as per SOP on CAPA (QA/021).
- If a batch or number of batches is affected, impact of Deviation and Incident shall be assessed on product quality before releasing batch/batches to the market (stability should be monitored, if required.
- Any critical / major Deviation and Incident in products meant for regulatory market shall be communicated to Regulatory affairs. Regulatory affairs shall further communicate the critical /major Deviation and Incident to the Regulatory authority / customer.
- The customer shall evaluate the deviation / incident and after study the impact shall report back to Regulatory affairs. Regulatory affairs shall communicate to unit Quality Assurance by email or fax.
- All the email/fax/communication related to the Deviation and Incident shall be attached to the deviation report.
- Deviation and Incident close out date shall be entered on the Deviation and Incident report by unit quality assurance with signature.
- Product related Deviation and Incident shall be reviewed and corrective actions shall be ensured by quality assurance before the dispatch of the batch.
- A corrective / preventive action shall be initiated for the repetitive Deviation and Incident in the SOP no. QA/021. A permanent change can be implemented as per the SOP of standard operating procedure for the handling ofchange control (SOP No. QA/017).
- CLOSURE OF DEVIATION AND INCIDENT:
- After the post implementation evaluation of Deviation and Incident implemented, QA Executive / designee shall do the closure of the Deviation and Incident form after.
- Ensuring results and supporting data.
- Completion of the required activities /documents identified during the evaluation and approval.
- Completion of Reference CAPA and Change control (if required).
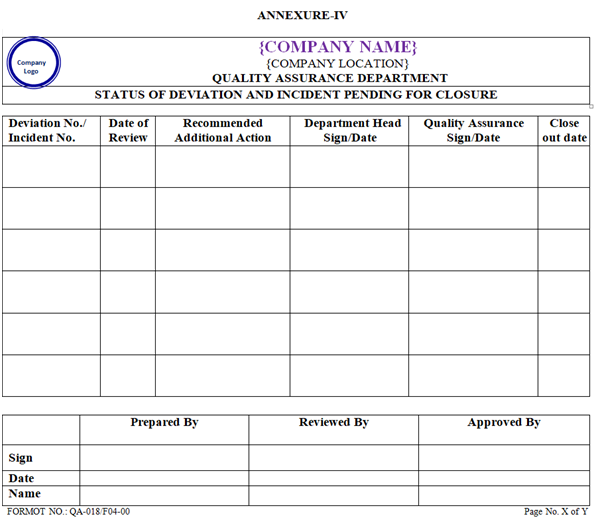
- Periodic quarterly review shall be performed by QA during March, June, September and December of each year to check for the closure of the Deviation and Incident, which is waiting for closure. Status of Deviation and Incident pending for closure shall be prepared by QA as per “Annexure-IV.
- All activities pertaining to the implementation of Deviation and Incident shall be completed within 180 working days from the date of approval of Deviation and Incident. If activity is not completed within given timeline then extension can be granted for maximum 90 days. Maximum 2 extensions can be taken. If after 2 extensions task is not closed then based on head QA recommendation, remaining task can be track with separate CAPA.
- Handling of Deviation and Incident shall be as per the flow chart:
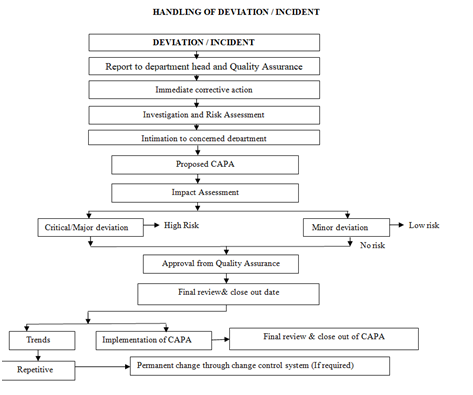
Click the link for download word file copy of this document: https://pharmaguidehub.com/product/standard-operating-procedure-for-the-handling-of-deviation-and-incident/
- REFERENCE:
21 CFR 211.100
- ANNEXURES:
| ANNEXURE No. | TITLE OF ANNEXURE | FORMAT No. |
| Annexure-I | Deviation and Incident Log Book | QA-018/F01-00 |
| Annexure-II | Deviation and Incident Report | QA-018/F02-00 |
| Annexure-III | Trend analysis for Incident/Deviation | QA-018/F03-00 |
| Annexure-IV | Status of Deviation and Incident pending for closure | QA-018/F04-00 |
| Annexure-V | Interview cum counseling form | QA-018/F05-00 |
| Annexure-VI | Action plan for Deviation and Incident based on trends | QA-018/F06-00 |
ENCLOSURES: SOP Training Record.
- DISTRIBUTION:
- Controlled Copy No. 01 : Head Quality Assurance
- Controlled Copy No. 02 : Head Quality Control
- Controlled Copy No. 03 : Head Production
- Controlled Copy No. 04 : Head Warehouse
- Controlled Copy No. 05 : Head Engineering
- Controlled Copy No. 06 : Head human resources
- Master Copy : Quality Assurance Department
- ABBREVIATIONS:
| SOP | : | Standard Operating Procedure |
| R & D | : | Research & development |
| SQC | : | Statistical Quality Control |
| CAPA | : | Corrective and Preventive Action |
| Dept. | : | Department |
| Mfg. | : | Manufacturing |
| Exp. | : | Expiry |
REVISION HISTORY:
CHANGE HISTORY LOG
| Revision No. | Details of Changes | Reason for Change | Effective Date |
| 00 | New SOP | Not Applicable | To be written manual |
Click the link for download word file copy of this document: https://pharmaguidehub.com/product/standard-operating-procedure-for-the-handling-of-deviation-and-incident/
ANNEXURE-I
| DEVIATION AND INCIDENT LOG BOOK |
| S. No. | Date of Issuance | Deviation No./ Incident No. | Concerned Department | Product / Item /Document / Equipment / Others | B.No. /A.R. No. / Code No. / Document No. / Others | Category (Critical/ Major / Minor) | Description of Deviation/ incident | Target Date completion | Actual Date of Compliance | CAPA Verified By QA (Sign & Date) | Deviation/ Incident Closed By (Sign & Date) | Remarks |
| Deviation and Incident Report No : |
| Any Previous Deviation / Incident of similar nature reported ( in previous one year) : Yes /No (Brief Description if answer is yes) Sign / Date Quality Assurance |
| Proposed Corrective and Preventive Actions: (To be filled by concerned Department) Sign/Date Sign / Date Department Head/Designee Quality Assurance |
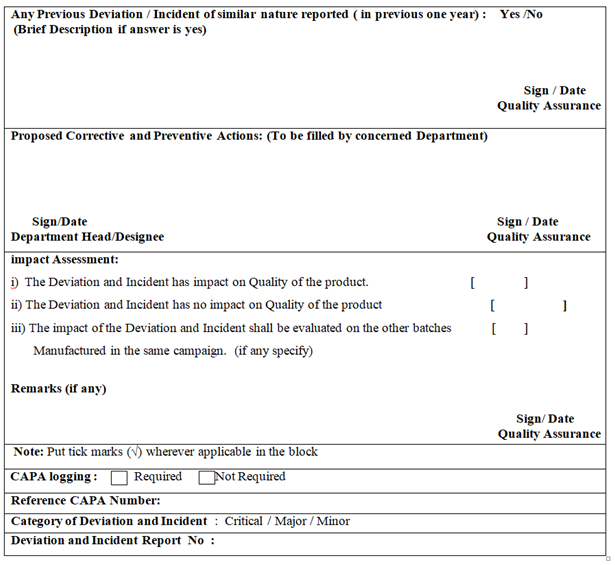
| impact Assessment: i) The Deviation and Incident has impact on Quality of the product. [ ] ii) The Deviation and Incident has no impact on Quality of the product [ ] iii) The impact of the Deviation and Incident shall be evaluated on the other batches [ ] Manufactured in the same campaign. (if any specify) Remarks (if any) Sign/ Date Quality Assurance |
| Note: Put tick marks (√) wherever applicable in the block |
| Reference CAPA Number: |
| Category of Deviation and Incident : Critical / Major / Minor |
| Deviation and Incident Report No : |
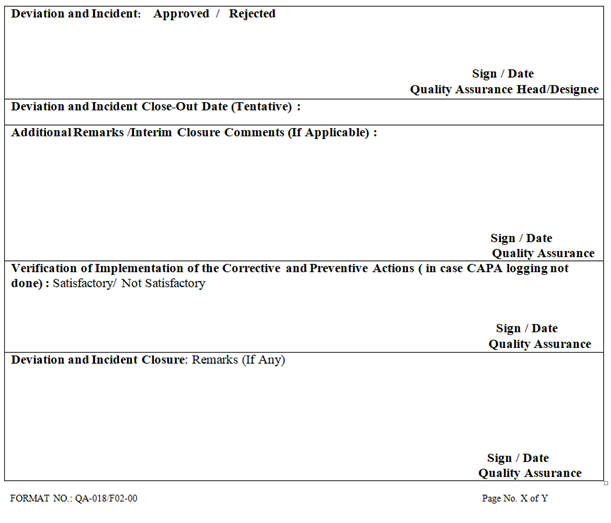
| Deviation and Incident: Approved / Rejected Sign / Date Quality Assurance Head/Designee |
| Deviation and Incident Close-Out Date (Tentative) : |
| Additional Remarks /Interim Closure Comments (If Applicable) : Sign / Date Quality Assurance |
| Verification of Implementation of the Corrective and Preventive Actions ( in case CAPA logging not done) : Satisfactory/ Not Satisfactory Sign / Date Quality Assurance |
| Deviation and Incident Closure: Remarks (If Any) Sign / Date Quality Assurance |
ANNEXURE-II
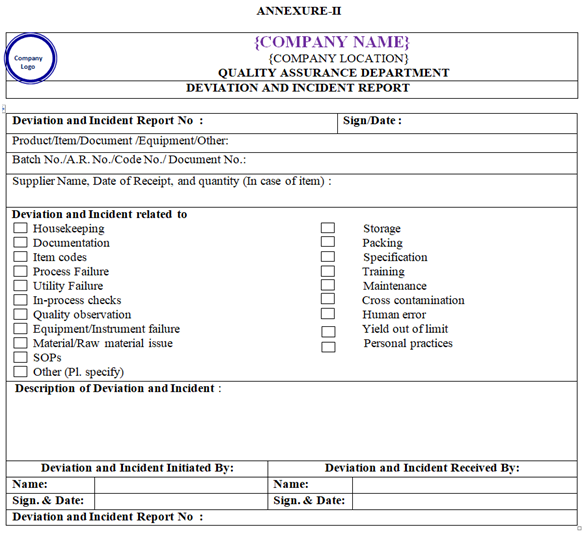

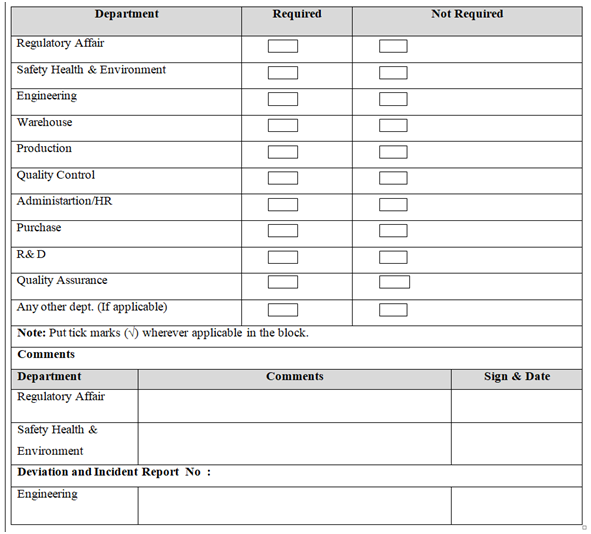

Click the link for download word file copy of this document: https://pharmaguidehub.com/product/standard-operating-procedure-for-the-handling-of-deviation-and-incident/
ANNEXURE-III

ANNEXURE-IV
ANNEXURE – V

ANNEXURE – VI

Click the link for download word file copy of this document: https://pharmaguidehub.com/product/standard-operating-procedure-for-the-handling-of-deviation-and-incident/


