
- OBJECTIVE:
To lay down a procedure that describes the approach for managing the risk of cross contamination.
- SCOPE:
This procedure is applicable for managing the risk of cross contamination at {Company Name} {Location}.
- RESPONSIBILITY:
- Production: To initiate the change control for introduction of new Active Pharmaceutical Ingredient (API) and finished drug product in the facility. To participate in the QRM exercise, wherever necessary. To participate in the execution of action items for control of cross contamination, wherever necessary. To communicate risks and risk reduction measures to the operations staff. To provide basic training to the operations staff on the control of cross-contamination.
- Quality Assurance: To review the change request related to new API and finished drug product
Introduction. To propose action items for control of cross contamination, wherever necessary. To review the assessment of cross contamination risk performed by the functional reviewers. To determine the requirement for conducting a formal QRM, wherever required. To document the requirement of the need for conducting formal or informal QRM in QA Comments field. To initiate formal QRM, wherever required. To ensure all changes to manufacturing facilities, manufacturing process, work procedures and product mix are evaluated for their impact on the control of cross-contamination. To ensure that the impact of any risk reduction measures are not detrimental to the systems, facilities, utilities, equipment or processes.
- ACCOUNTABILITY:
QA Head shall be Accountable for implementation of SOP.
- PROCEDURE:
- A formal assessment of the control measures required to accompany new Active Pharmaceutical Ingredient (API) and finished drug product shall be conducted prior to introduction of the material and product into the product matrix of an existing manufacturing operations.
- All existing facilities should conduct an assessment of the current operations.
- A formal assessment of the controls required to accompany existing compounds shall be integrated into the design of all new manufacturing operations.
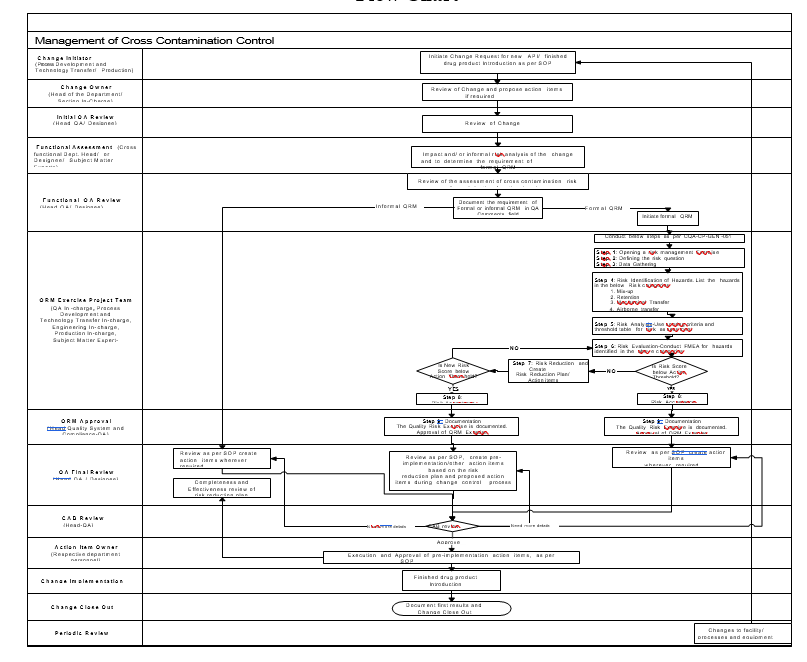
- The change management process of any new API/finished drug product introduction into the facility described in the procedure shall also consider the assessment of risk of cross contamination as below.
- The change initiator shall initiate the change request for any new API/finished drug product introduction into the facility. The change request shall be reviewed by the change owner and relevant action items shall be proposed for cross contamination control, in addition to the other action items for introduction of new API/finished drug product such as cleaning method verification, stability protocol, batch manufacturing record etc.
- Further QA reviewer shall also review the change request and propose the action items for control of cross contamination in addition to other action items for introduction of new API/finished drug product such as cleaning method verification, stability protocol, batch manufacturing record etc.
- The relevant Cross Functional Department Head/or Designee/Designated Subject Matter Experts (SME) from various disciplines shall also assess the change request for introduction of new API/finished drug product for its impact on risk of cross contamination through informal risk analysis or identifying the requirement for a formal QRM.
- Based on the functional QA review, the QA reviewer shall determine the need of formal or informal QRM exercise for risk of cross contamination due to introduction of new API/finished drug product and document the same in the ‘QA Comments’ field in the change request form.
- If a formal risk assessment is not required based on above step, an informal risk assessment shall be performed as per the procedure mentioned in SOP.
- Subsequently the change request shall be reviewed and approved for implementation as per the SOP.
- If a formal QRM exercise is required, the exercise shall be initiated.
- The formal quality risk assessment undertaken to control the risk of cross-contamination shall be scientific and risk-based process as detailed below.
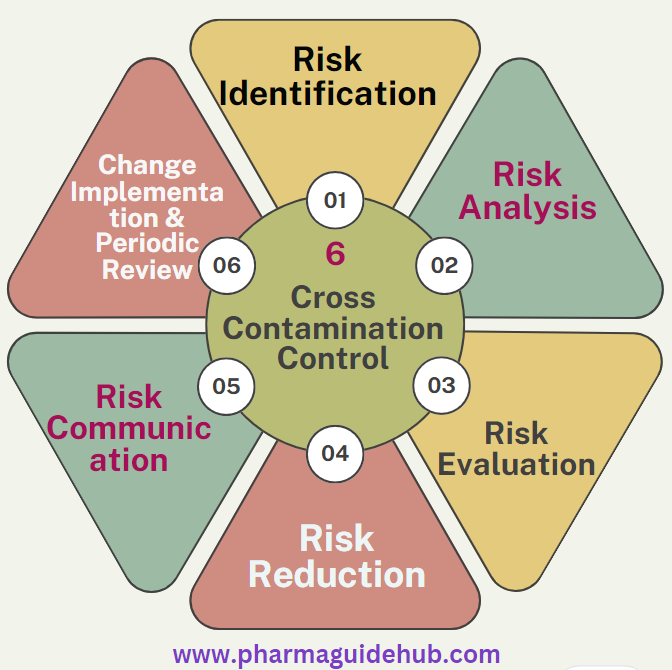
- Risk Identification:
- The Allowable Daily Exposure (ADE) or Permissible Daily Exposure (PDE) value of the API generated by toxicologists shall be the basis for performing QRM exercise for assessing the risk of cross contamination.The Thresholds of Toxicological Concern Concept as per SOP shall be adopted in case of non-availability of ADE/PDE values.If an ADE/PDE is assigned using the Thresholds of Toxicological Concern Concept, it shall be periodically reviewed at pre-defined intervals to determine if the assignment is still valid. When there is ADE/PDE data available, the same shall be updated to perform risk evaluation of cross contamination.
- The following pre-existing information, but not limited to, shall be collected prior to performing risk analysis:
Click the link for download word file copy of this document: https://pharmaguidehub.com/product/cross-contamination-control-in-pharmaceutical/
- Cleaning validation impact assessment document.
- List of compounds to be processed (including corresponding ADE/PDE).
- Facility drawings including HVAC zoning and pressure cascades.
- Flow diagrams for people, products, wastes and equipment.
- Process descriptions – including process equipment information.
- Historical data – including process nonconformance reports, recalls, customer complaints, failure investigations, cleaning data, other test data such as gradient studies
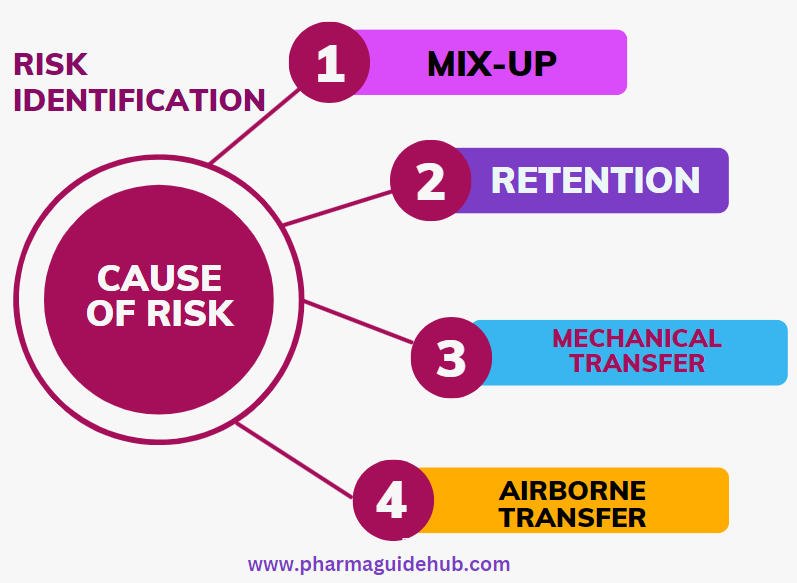
- The risk identification shall consider the four possible routes of potential exposure of cross contamination i.e. mix-up, retention, mechanical transfer and airborne transfer shall be assessed.
- Mix-up may occur when any of the following items occur:
- The risk identification shall consider the four possible routes of potential exposure of cross contamination i.e. mix-up, retention, mechanical transfer and airborne transfer shall be assessed.

Click the link for download word file copy of this document: https://pharmaguidehub.com/product/cross-contamination-control-in-pharmaceutical/
- The wrong API is used in the process
- Dirty equipment is used instead of clean equipment
- The wrong dedicated part is used
- The wrong label is placed on the container
- To assess the risk of mix-up, the above factors shall be considered during the review of flow diagrams of personnel, materials, and equipment and waste streams. In particular, care must be given to those areas where multiple flows intersect or overlap.
- Retention may occur when any of the following items occur:
- Cleaning procedure cannot meet Acceptance limit. Scientifically justifiable health-based cleaning limits are developed using the ADE/PDE as the basis.
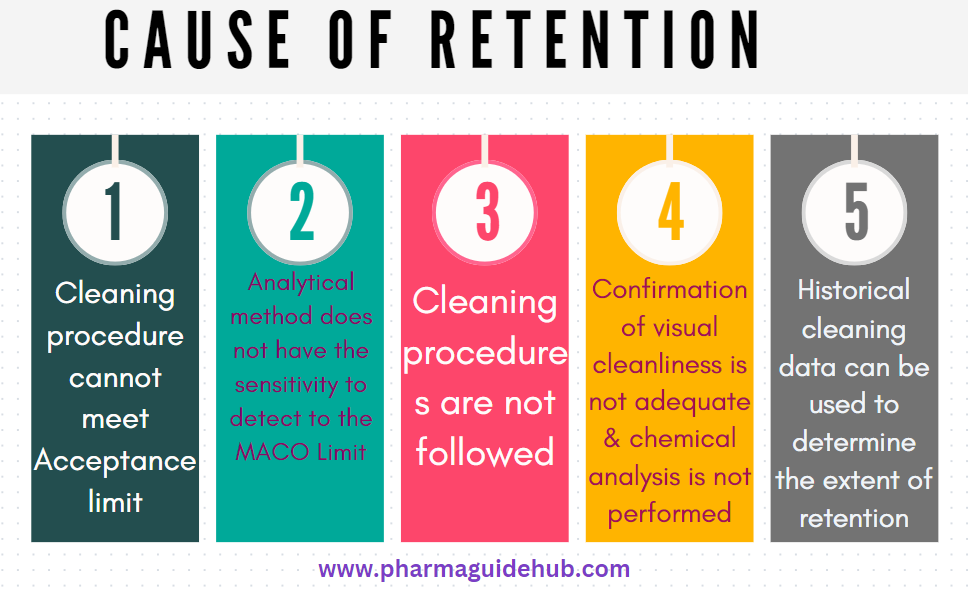
- Analytical method does not have the sensitivity to detect to the MACO Limit
- Cleaning procedures are not followed
- Confirmation of visual cleanliness is not adequate (the required limit is below the visual detection limit) and chemical analysis is not performed.
- To assess the risk of retention, historical cleaning data can be used to determine the extent of retention or carry over in a process.Mechanical transfer may occur when particulate is carried from one process area into another via portable equipment, personnel, gowning, containers and wastes. The transfer procedures for material, equipment, and waste materials through the facility shall be reviewed for assessing the possibility of cross contamination. The existing control measures shall also be reviewed including the removal of surface residues on items entering the common area through wiping, gowning procedures when exiting a process room into a common area etc. Airborne transfer may occur when an airborne aerosol from one process sediments onto non-product contact surfaces and gets re-entrained into the airstream and is deposited into another process. To assess the risk of airborne transfer, the pressure cascades, airlocks and/or filtration systems shall be considered during risk analysis.
- Risk Analysis:
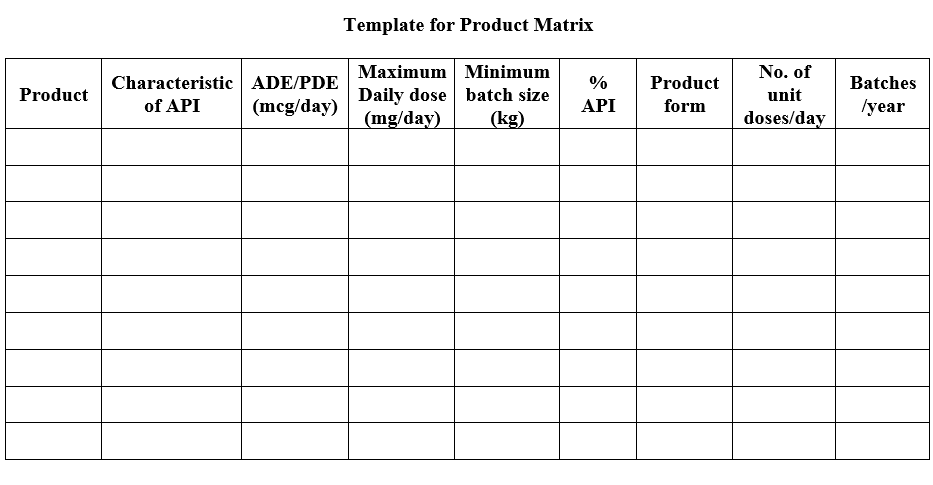
- A Product Matrix shall be developed to determine the greatest risk API and the most vulnerable product for the facilities/block. The matrix must incorporate the hazard and characteristics of the API, volumes manufactured frequency of manufacture, and processes used.
- The Product Matrix data shall be used for conducting the Failure Mode and Effects Analysis (FMEA).
- Key stakeholder members representing Quality Assurance, Production and Engineering at a minimum shall participate in the FMEA session.
- The following scoring shall be used for performing FMEA exercise:
- Risk Analysis:
| Score | Severity (ADE mcg/day) | Occurrence | Detection | |
| Time based | Batch based | |||
| 10 | ≤ 10 | Once or more per day | Once or more per batch | Cannot be detected |
| 7 | 11-110 | Once or more per month | 50% or more of batches | Manual based system |
| 5 | 101 – 1000 | Once or more per year | 10 – 49% of batches | Inspection of statistically based sampling |
| 3 | 1000 to 9999 | Greater than once per year | < 10% of batches | Detected by analysis |
| 1 | ≥ 10,000 | Greater than once per 5 years | < 1% of batches | Obvious or automatically alarmed |
- Risk Evaluation:
- A risk score shall be obtained for each identified and assessed failure mode and the risk score shall be compared to the Action Threshold as outlined in the following table.
| RPN Number | Ranking | Production | Investigation | Remedial Action | Review |
| 45 – 75 | Unacceptable Risk | Cease | Yes | Yes | Monthly once remediated |
| 15 – 44 | High risk | Continue | Yes | Yes | Monthly |
| 10 – 14 | Significant Risk | Continue | Yes | Yes | Six Monthly |
| 1 – 9 | Acceptable Risk | Continue | No | No | Annually |
- Incase if the risk score is below the action threshold, the QRM shall be approved and documented.
- If the risk score is above the action threshold, risk reduction measures shall be identified to mitigate the risk of cross contamination.
Click the link for download word file copy of this document: https://pharmaguidehub.com/product/cross-contamination-control-in-pharmaceutical/
- Risk Reduction:

- All identified risks that require remediation (as outlined above), shall be created as pre implementation action items as per the SOP. Necessary CAPAs shall be implemented and verified to reduce the RPN values.
- If the risk is still not acceptable, additional risk reduction measures shall be implemented and verified as above. This cycle continues until the risks are deemed acceptable.As part of the Risk Reduction plan, control measures shall be identified to mitigate the cross-contamination risk.
- All items that are deemed acceptable risk will be monitored/reviewed annually to ensure that the risk remains acceptable.All control measures must be accompanied by mechanisms designed to detect breaches to the systems in place. As such, audible, visual or otherwise recorded and monitored alarms must be utilized where feasible or implementation verification of procedure, to assist in reducing risks to an acceptable level.Whenever possible, methods to increase detection should be incorporated into systems that manage the risk of cross-contamination.Wherever required, the pressure cascades, airlocks and/or filtration system should be enhanced to mitigate the risk of cross-contamination.
- Based on the risk reduction measures, the risk shall be re-evaluated and if the risk score is below the action threshold, the QRM shall be approved and documented.
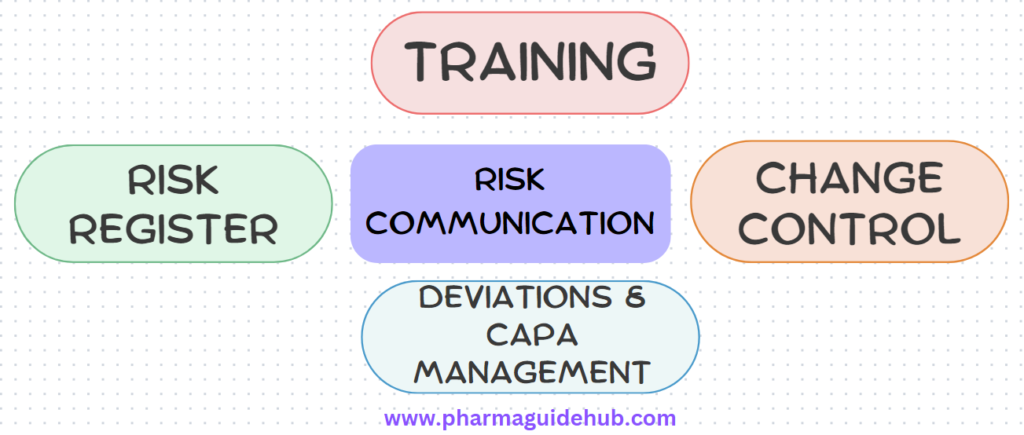
- Risk Communication:
Click the link for download word file copy of this document: https://pharmaguidehub.com/product/cross-contamination-control-in-pharmaceutical/
- Risk communication must be incorporated into the following quality systems:
- Training
- Change Control
- Deviations and CAPA Management
- Risk Register
- The outcome of the QRM shall be communicated in an iterative and continual manner to all the stakeholders.
- Change Implementation:
- The proposed change shall be evaluated by the Change Control Coordinator and the action items created at different stages of the approval process shall be reviewed. Based on the evaluation, Change Control Coordinator shall create or delete action items.
- The proposed change shall be reviewed and approved by the Quality Assurance Head.
- Action items shall be generated to designated owners in the following order based on the action item type selected during creation of action items.
- Pre-Implementation action items
- Implementation action items
- Follow-up action items
- All pre-implementation action items shall be executed by the action item owner and upon satisfactory implementation, action items shall be approved by the action item approver as per the SOP.
- After execution and verification of pre-implementation action items, the new finished drug product can be introduced.
- PeriodicReview:
- A review of the QRM exercise for cross contamination and risk profile of the facility must occur when any of the following occur:
- When there are changes to the facility, equipment and/or processes where the impact assessment indicates the requirement to review QRM.
- Update to SOPs for gowning, material transiting the facility, cleaning, filter certification and replacement.
- When new products are introduced to the facility where the impact assessment indicates the requirement to review QRM.
- When any of the systems in place to manage the risk of cross contamination is shown to be out of control (process non-conformances, out of specifications, breach in containment, etc.)
- Further, a review of the QRM and facility risk profile must be undertaken as per the step ‘Risk Evaluation’ based on the risk ranking to evaluate the effectiveness of the risk reduction measures. The review frequency can be reduced if adequate data is generated and risk ranking is determined to be low.
- Minimally, a review of the QRM and facility risk profile must be undertaken during the product quality review if none of the above events have been documented to have occurred.
Click the link for download word file copy of this document: https://pharmaguidehub.com/product/cross-contamination-control-in-pharmaceutical/
- REFERENCES:
Not Applicable
- ANNEXURES:
Not Applicable
ENCLOSURES: SOP Training Record.
- DISTRIBUTION:
- Controlled Copy No. 01 : Head Quality Assurance
- Controlled Copy No. 02 : Head Production
- Controlled Copy No. 03 : Head Warehouse
Master Copy : Quality Assurance Department
- ABBREVIATIONS:
| RPN | : | Risk Priority Number |
| No. | : | Number |
| SOP | : | Standard Operating Procedure |
| CAPA | : | Corrective And Preventive Actions |
| QRM | : | Quality Risk Management |
| ADE | : | Acceptable Daily Exposure |
| FEMA | : | Failure Mode and Effect Analysis |
| API | : | Active Pharmaceutical Ingredient |
- REVISION HISTORY:
CHANGE HISTORY LOG
| Revision No. | Details of Changes | Reason for Change | Effective Date |
| 00 | New SOP | Not Applicable | To be written manual |
Click the link for download word file copy of this document: https://pharmaguidehub.com/product/cross-contamination-control-in-pharmaceutical/
Flow Chart
Template for Product Matrix
Template for FMEA


Frequently Asked Questions?
Q: What is the purpose of conducting a formal assessment of control measures for new API and finished drug product introduction in existing manufacturing operations?
A: The purpose of conducting a formal assessment is to evaluate and ensure the adequacy of control measures accompanying the introduction of new Active Pharmaceutical Ingredients (API) and finished drug products into the existing manufacturing operations. This assessment is crucial for identifying and managing potential risks, including cross-contamination, prior to integrating the new material and product into the product matrix of the existing operations.
Q: How is the change management process related to the assessment of the risk of cross-contamination during the introduction of new API/finished drug products?
A: The change management process is intricately linked to the assessment of the risk of cross-contamination. When initiating a change request for the introduction of new API/finished drug products, the change initiator initiates the change request, which undergoes reviews by the change owner, QA reviewer, and Cross Functional Department Head or designated Subject Matter Experts (SMEs). The assessment includes action items specifically addressing cross-contamination control, in addition to other action items like cleaning method verification, stability protocol, and batch manufacturing record.
Q: How is the need for a formal or informal Quality Risk Management (QRM) exercise determined in the context of cross-contamination risk assessment?
A: The need for a formal or informal QRM exercise is determined through a series of steps. The QA reviewer, based on functional review, decides whether a formal QRM exercise is necessary. If not, an informal risk assessment is performed following the procedure mentioned in the SOP. The decision is documented in the ‘QA Comments’ field in the change request form. If a formal QRM exercise is required, it is initiated to scientifically and risk-basedly control the risk of cross-contamination.
Q: Can you explain the key steps involved in the formal quality risk assessment undertaken to control the risk of cross-contamination?
A:
- Risk Identification :
- Based on Allowable Daily Exposure (ADE) or Permissible Daily Exposure (PDE) values or Thresholds of Toxicological Concern.
- Collection of pre-existing information such as cleaning validation impact assessment, list of compounds, facility drawings, process descriptions, and historical data.
- Risk Analysis :
- Development of a Product Matrix to determine the greatest risk API and the most vulnerable product.
- Conducting Failure Mode and Effects Analysis (FMEA) using data from the Product Matrix.
- Scoring severity, occurrence, and detection for FMEA.
- Risk Evaluation :
- Comparison of risk score with Action Threshold to determine risk acceptability.
- Approval and documentation of QRM if the risk score is below the action threshold.
- Identification of risk reduction measures if the score is above the threshold.
Q: What is the role of Risk Priority Number (RPN) in the risk evaluation process?
A: The Risk Priority Number (RPN) is calculated by multiplying the severity, occurrence, and detection scores during the risk analysis . The RPN provides a numerical value that represents the overall risk associated with a specific failure mode. In risk evaluation , the RPN is compared to the Action Threshold to determine the acceptability of risk. Different ranges of RPN numbers are associated with specific risk rankings, guiding decisions on whether to cease, continue with additional actions, or accept the risk.
Q: How is risk communication integrated into the overall quality systems after the risk evaluation process?
A: Risk communication is incorporated into various quality systems, including training, change control, deviations and CAPA management, and the risk register. The outcome of the Quality Risk Management (QRM) process is communicated iteratively and continually to all stakeholders. This ensures that relevant information regarding cross-contamination risks is disseminated effectively across different aspects of the quality management system.
Q: What steps are involved in the change implementation process after the risk assessment and reduction measures have been determined?
A:
- Evaluation and Approval :
- Evaluation of proposed changes by the Change Control Coordinator.
- Review and approval by the Quality Assurance Head.
- Action Items Generation :
- Generation of action items for designated owners based on the type of action item (pre-implementation, implementation, follow-up).
- Execution and Verification :
- Execution of pre-implementation action items by the owner.
- Approval of action items upon satisfactory implementation.
- Introduction of New Finished Drug Product :
- After execution and verification of pre-implementation action items, the new finished drug product can be introduced.
Q: Why is a periodic review of the Quality Risk Management (QRM) exercise and facility risk profile necessary, and what events trigger this review?
A: A periodic review of the QRM exercise and facility risk profile is necessary to ensure ongoing effectiveness and relevance. Trigger events for this review include changes to the facility, equipment, or processes, updates to relevant SOPs, introduction of new products, and instances where systems in place to manage cross-contamination risk show signs of being out of control (process non-conformances, out-of-specifications, breaches in containment, etc.). Additionally, the review frequency can be adjusted based on data generation and risk ranking results. Minimally, a review is undertaken during product quality reviews if none of the specified events have occurred.
Click the link for download word file copy of this document: https://pharmaguidehub.com/product/cross-contamination-control-in-pharmaceutical/

