

- OBJECTIVE:
The objective is to define the procedure to be followed for initiating and handling of the NDA-Field Alert Report.
- SCOPE:
This Standard Operating Procedure is applicable to all the US distributed products for the ANDA application holder.
- RESPONSIBILITY:
- QA: shall receive the notification of event and escalate the same to QA Head & Compliance In-charge (QA) respectively. Evaluate the necessity to file Field Alert Report in co-ordination with QA head & In-charge (QA). Draft the FAR in co-ordination with QA head & Compliance In-Charge for approval. Monitor the progress of the Field Alert
- ACCOUNTABILITY:
QA Head shall be Accountable for implementation of SOP.
- PROCEDURE:
- NDA-Field Alert Report (FAR) shall be initiated in the event of any issue that can impact the quality of distributed product of one batch or other associated batches for factors such as given but not limited to:
- Any incident that causes the drug product or its labeling to be mistaken for, or applied to, another article (adulterated or misbranded). E.g. Wrong product (label and contents are different products); Correct product but wrong strength; Missing label, Mislabeling i.e. wrong or missing text or figures; Missing or incorrect information (leaflets or inserts) etc.
- Significant chemical, physical or other change or deterioration in the distributed drug product. E.g. Product discoloration, Unknown spots on product, Significant impurities, Cross contamination, Particulates, Foreign contamination, Uncrimped/partially Crimped vial seal, No/partial lyophilization cake, Melt back etc.
- Failure of one or more distributed batches to meet the established specifications. E.g. Difference in product Appearance, Dissolution failures, Stability failures etc.
- Confirmation of bacterial contamination in the distributed drug product.
- Information regarding any distributed product to be the subject of counterfeit activity that may warrant criminal enforcement.
- Field Alert Report is required for the above mentioned factors wherein the following actions are identified to be necessary
Click the link for download word file copy of this document: https://pharmaguidehub.com/product/field-alert-reporting/
- Further investigation is required
- Product Recall, if any.
- Field Alert Report is not required for an event in case,
- If the product has not been distributed.
- Reported events are invalidated within 3 US National working days.
- The reporting time starts when the firm (PGH or Distributor) becomes aware of a reportable event by following means,
- Verbal or written complaint
- Internal testing
- Unconfirmed Observation
- Confirmed Observation
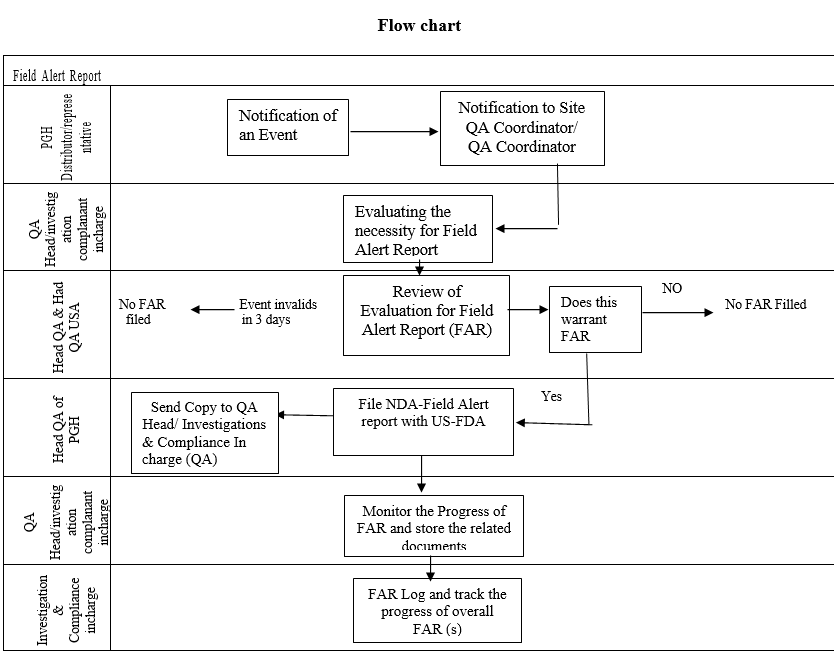
- If the event is reported to an PGH Representative or Distributor, the same shall be notified to the respective QA Co-Ordinator/Compliance complaints Co-ordinator of PGH within one (1) US National Working Day.
- The rationale for filing or not filing a Field Alert shall be recorded in the Evaluation form for Field Alert Reporting by the QA Head in coordination with Investigations & Compliance –In Charge (QA).
- The Evaluation Form for Field Alert Reporting shall be reviewed and approved by Head QA with appropriate comments.
- The same shall be submitted to the Head-QA of PGH USA electronically for concurrence and further courses of action.
- Head-QA of PGH USA shall approve the Evaluation Form for Field Alert Reporting and shall send to Investigations & Compliance In-Charge (QA) and QA Head for record.
- The approved Evaluation Form for Field Alert Reporting shall be filed along with the respective Interim/Follow Up/Final Investigation Report, as applicable.
- Based on the conclusion of the evaluation, a Field Alert shall be filed for an event, as applicable.
- A decision for filing Field Alert shall be concluded and the Field Alert filed within three (3) US National working days from the notification of the discrepancy/receipt of the information by any PGH Employee/Representative or Distributor.
If multiple NDAs or ANDAs are involved, submit one Form FDA 3331a for individual NDA/ANDA. For multiple unrelated issues, separate FARs shall be submitted for each issue type.
- The Initial FAR shall be submitted to Head-QA of PGH USA for review.
- FAR shall be reviewed by the Head-QA of PGH USA and filed with the US-FDA.
- Investigations & Compliance complaints coordinator shall closely monitor the filing of FAR by Head-QA of PGH USA on a daily basis.
- If the filed field alert report is not received on the same day of filing, Investigations & Compliance complaints coordinator shall alert the Head-QA of PGH USA for non-receipt of filed FAR.
- If the FAR is not filed with FDA by Head-QA of PGH USA within 3 US National Working days, Investigations & Compliance complaints coordinator shall alert the Head-QA of PGH USA and ensure that the FAR shall be filed within 1 day from the identification of discrepancy.
- A Process Non Conformance shall be initiated by Head-QA of PGH USA if FAR is not filed within 3 US National Working days, after notification of FAR.
Click the link for download word file copy of this document: https://pharmaguidehub.com/product/field-alert-reporting/
If investigation of an observed Out of Specification (OOS) result on a distributed batch is ongoing, an Initial FAR shall be submitted within 3 US National Working days of observation. In the event the OOS result is invalid based on the conclusion of the investigation, a Follow-up and/or a Final FAR shall be submitted to the US-FDA
- The FAR can be communicated by telephone and shall be followed by a written Report (FAR) through communication means such as facsimile or e-mail or courier to the NDA- Field Alert Report coordinator in the concerned jurisdictional US FDA District Office.
- The FAR and its mailing cover should be plainly marked: “NDA -Field Alert Report”.
- A Follow-up FAR shall be submitted to US-FDA in case if any new information/data or some conclusion is developed based on the ongoing investigation.
Preferably, the Follow-up FAR shall be submitted to US-FDA within 5 days from learning a new information/data or a conclusion is developed
- If the investigation is completed, a Final FAR shall be filed to close out the case.
- The Head QA of PGH USA shall send a copy of the filed FAR and the same shall be filed in the respective files by the QA Officer/Designee.
- The investigation concerning the Field Alert shall be completed within lead time as per respective event SOP/Failure Investigation SOP. In case of any extension requirement, the corresponding SOP shall be followed.
- Wherever applicable, Product Recall shall be initiated as per the corresponding SOP.
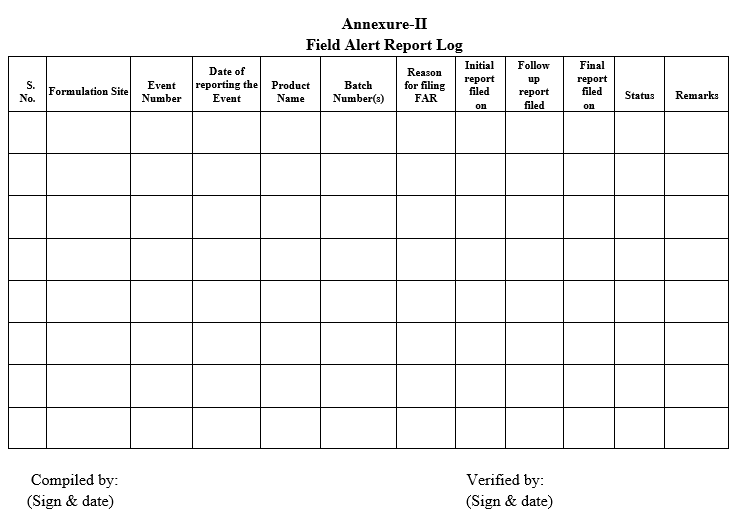
- Copy of all documents, supporting data and communications shall be stored as a separate docket along with the FAR with the respective QA. Investigations & Compliance In-charge/designee (QA) shall maintain an active spread sheet with all the FAR and their progress. The status of the same shall be reviewed on a monthly basis and shall be retained as a controlled document.
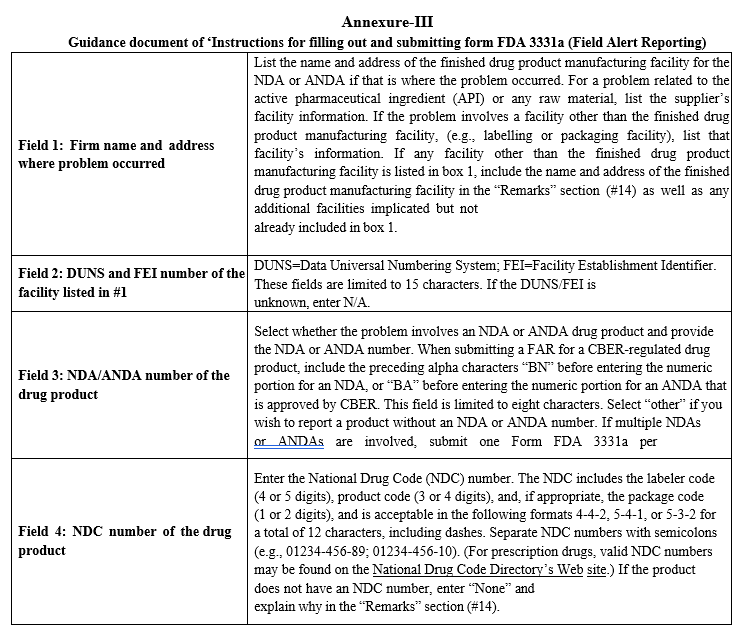
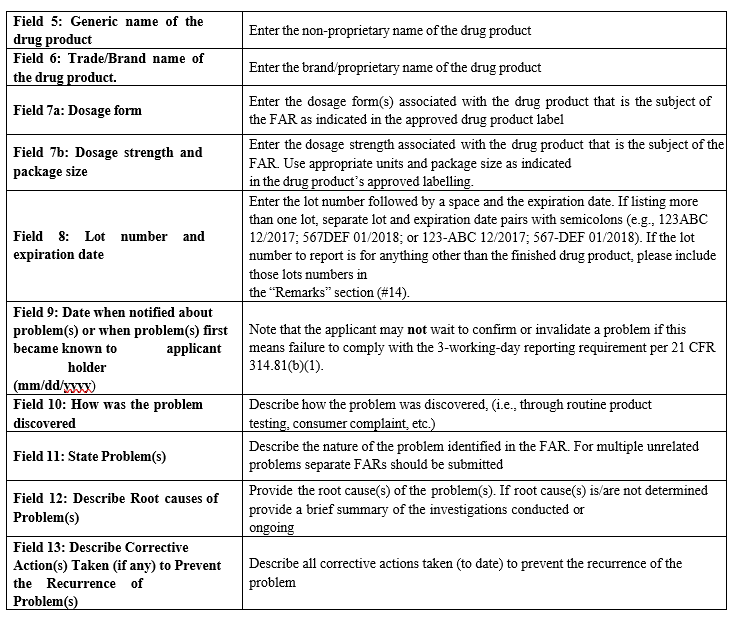
- Filling and Submission of Field Alert Report to FDA:Field Alert Report Form (Form FDA 3331a) shall be obtained by accessing the below link from FDA Website : https://www.fda.gov/downloads/AboutFDA/ReportsManualsForms/Forms/UCM350286.pdf The form FDA 3331a (Field Alert Reporting) shall be filed as per ‘Instructions for filling out and submitting form FDA 3331a. QA Head in coordination with Investigations & Compliance Co-ordinator shall fill the details in editable FAR form (FDA 3331a Form) and submit the FAR copy (PDF filled editable copy) to Head-QA of PGH USA for review and submission to FDA.Head-QA of PGH USA shall review the FAR copy and make corrections, if necessary generate the Read-Only version of FAR copy by;
- Printing and scanning the completed form to create a PDF file.
- Using the File/Print option to print the completed form as an Adobe PDF to create a Read-Only version of the form.
- After creating PDF Read-Only version of FAR copy, click the ‘Submitby Email’ option (available at the end of the FAR form) that opens a new Outlook email, addressed to respective ORA District Office based on the district office selected in the form.
- In the automatically generated mail, CDER-XML-FAR mailbox will be listed as a “Cc” recipient and an XML file generated by the form will be attached with a mail subject.
- The email subject line will be automatically generated with information from fields 3 (NDA/ANDA/Other Number) and 5 (Generic Name of the Product) in the 3331a form, with submission date and time stamp.
- The Read-Only Version of FAR copy shall be attached to this auto generated mail (attached with XML file) and the email along with XML and PDF FAR copies shall be sent to FDA without changing the mail subject line
- REFERENCES:
- 21 CFR Section 314.81 Other Post Marketing Reports
- For 3331a form – https://www.fda.gov/downloads/AboutFDA/ReportsManualsForms/Forms/ UCM350286.pdf
- ANNEXURES:
| ANNEXURE NO. | TITLE OF ANNEXURE |
| Annexure-I | Evaluation Form for Field Alert Reporting |
| Annexure-II | Field Alert Report Log |
| Annexure-III | Guidance document of ‘Instructions for filling out and submitting form FDA 3331a (Field Alert Reporting): |
- DISTRIBUTION:
- Controlled Copy No. 01 : Head Quality Assurance
- Master Copy : Quality Assurance Department
- ABBREVIATIONS:
| USFDA | : | United States Food and Drugs Administration |
| No. | : | Number |
| SOP | : | Standard Operating Procedure |
| OOS | : | Out of Specification |
| QRM | : | Quality Risk Management |
| FAR | : | Field Alert Report |
| XML | : | Extensible Mark-up Language |
| PGH | : | Pharmaguidehub |
| CFR | : | Code of Federal Regulations |
- REVISION HISTORY:
CHANGE HISTORY LOG
| Revision No. | Details of Changes | Reason for Change | Effective Date |
| 00 | New SOP | Not Applicable | To Be Written Manual |
Click the link for download word file copy of this document: https://pharmaguidehub.com/product/field-alert-reporting/
Flow chart
Annexure-I
Evaluation Form for Field Alert Reporting

Annexure-II
Field Alert Report Log
Annexure-III
Guidance document of ‘Instructions for filling out and submitting form FDA 3331a (Field Alert Reporting)

Click the link for download word file copy of this document: https://pharmaguidehub.com/product/field-alert-reporting/



