

- OBJECTIVE:
To lay down a procedure for good documentation practices requirements for the compliant, consistent and accurate completion of GxP Documentation.
- SCOPE:
This Procedure applies to all cGxP documents (electronic and handwritten) used in the manufacturing, packaging, labelling, testing, storage and distribution of Drug Products and Drug Substances for commercial distribution. It also applies to documents generated during Process Development, Formulation Development, Scale-Up Development, Analytical Method Development, Technology Transfer, Analytical Method Transfer and processing of Exhibit Batches in the general formulation facilities.
- RESPONSIBILITY:
QA Department shall be responsible for:
To maintain the document movement record.
To follow the procedure of Good Documentation Practice.
All User Departments / Personnel shall be responsible for:
All user department person who prepares the documents and records shall be responsible to follow the procedure mentioned herein
Quality assurance Head shall be responsible for:
To ensures & responsible for implementation of the defined system.
Plant Head:
To ensure implementation of the defined system.
- DEFINITIONS:
Good documentation practices (GDP): Good Documentation Practices are methods for recording, correcting and managing data, documents and records, to ensure the reliability and integrity of information and data throughout all aspects of a product’s lifecycle.
Good Documentation is a term used to describe standards by which documents are created, maintained, retained and dispositioned. Following Good Documentation Practices enables preparation of the documents and making entries of data in a legible and traceable manner.
GXP: Acronym for the group of good practice guides governing the preclinical, clinical, manufacturing, testing, storage, distribution and post-market activities for regulated pharmaceuticals, biological and medical devices, such as good laboratory practices, good clinical practices, good manufacturing practices, good pharmacovigilance practices and good distribution practices.
Document: A document is a physical or digital representation of a body of information designed with the capacity (and usually intent) to communicate intended subject. A document may manifest symbolic, diagrammatic or sensory representational information. For example, SOP, STP, Specifications, Master formula, Quality Policy, Protocols, etc.
Record: Any Document which contains recorded evidence of any GMP activity done during Manufacturing, testing, holding and release of a product. The record could be handwritten or electronically generated. Examples of records: Equipment/ Facility maintenance history, Change Control records, executed batch manufacturing record, Executed batch packaging record, Validation reports, stability reports, Executed Logbooks, Training records etc.
Click the link to download word file copy of this document: https://pharmaguidehub.com/product/gxp-practices-in-pharmaceuticals/
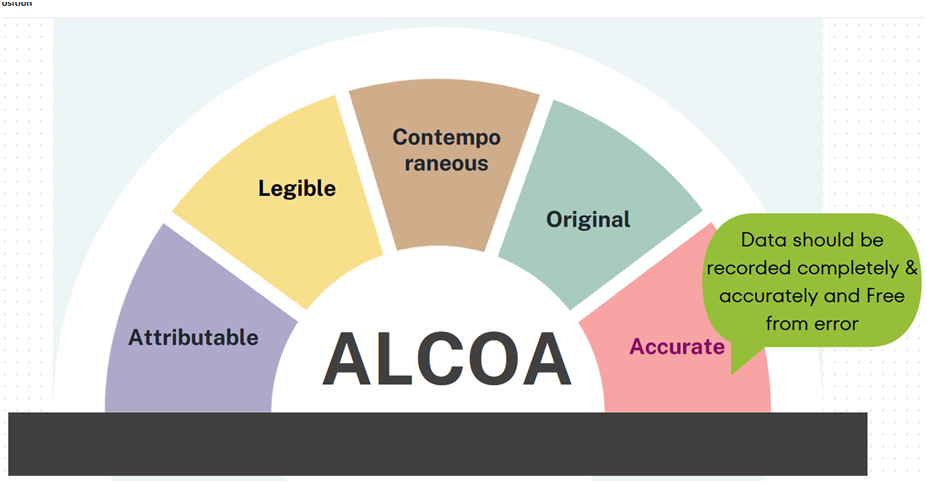
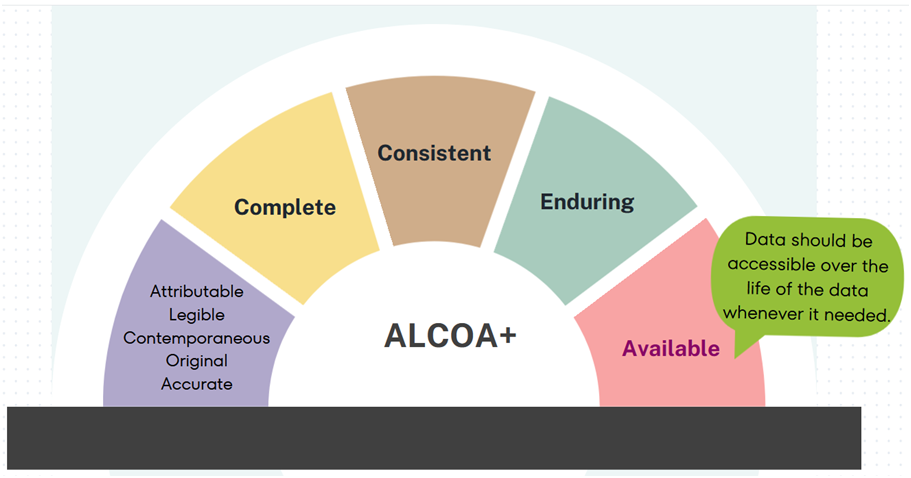
ALCOA: A commonly used acronym for ‘Attributable, Legible, Contemporaneous, Original and Accurate’.
Attributable: All data generated or collected must be attributable to the person generating the data. This should include whom performed an action and when. This can be recorded manually by initialling and dating a paper record or by audit trail in an electronic system.
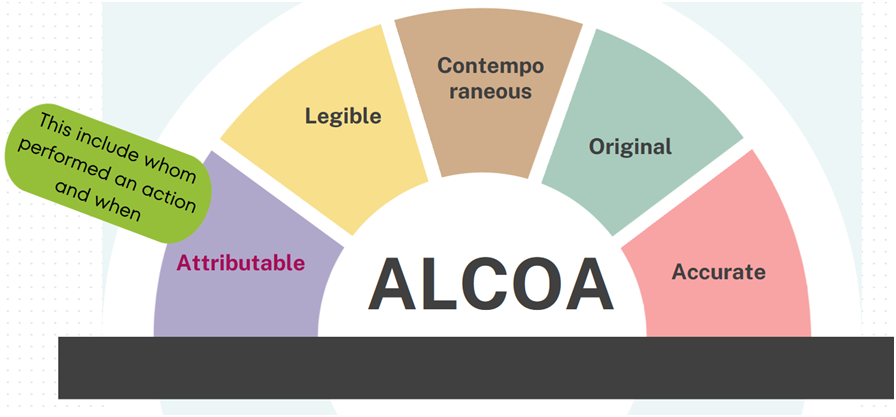
Note: It is important to ensure a signature log is maintained to identify the signatures, initials of people completing paper records.
For example:
During a validation exercise, test results should be initialed and dated by the person executing the test.
Adjustment of a set point on a process or monitoring system should be made by an authorised user and the details of the change logged in an audit trail.
A correction on a lab record should be initialed and dated to show when and who made the adjustment.
Legible: All data recorded must be legible (readable) and permanent. Ensuring records are readable and permanent assists with its accessibility throughout the data lifecycle. This includes the storage of human-readable metadata that may be recorded to support an electronic record.
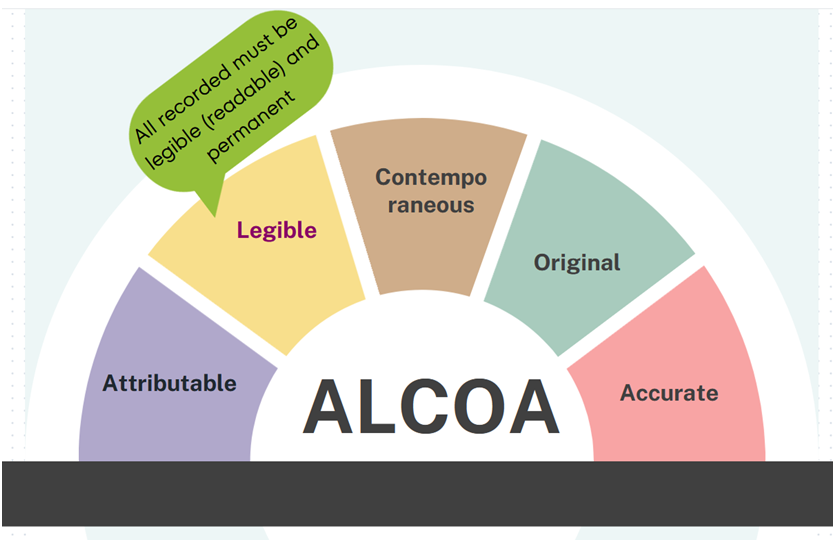
For example:
GDP will always promote the use of indelible ink when completing records.
When making corrections to a record, ensure a single line is used to strike out the old record. This ensures the record is still legible.
Controlling your paper records/forms and formatting them such that there is ample room for the information to be recorded.
Contemporaneous: Contemporaneous means to record the result, measurement or data at the time the work is performed. Date and time stamps should flow in order of execution for the data to be credible. Data should never be back dated.
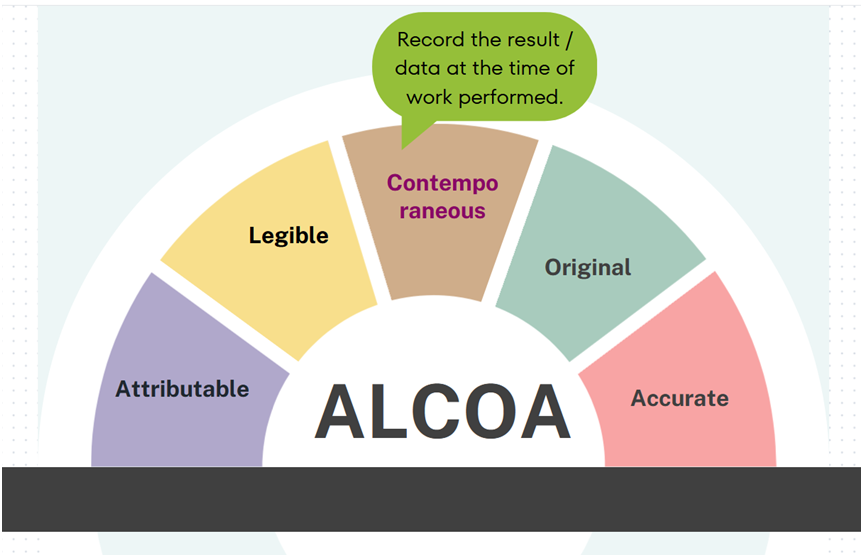
For example:
If executing a validation protocol, tests should be performed and their results recorded as they happen on the approved protocol.
Data that is logged, or testing that is performed electronically, should have a date/time stamp attached to the record.
Click the link to download word file copy of this document: https://pharmaguidehub.com/product/gxp-practices-in-pharmaceuticals/
Ensure electronic systems that log data have their system clocks synchronized.
Original: Original data, sometimes referred to as source data or primary data, is the medium in which the data point is recorded for the first time. This could be a database, an approved protocol or form. It is important to understand where your original data will be generated so that its content and meaning are preserved.
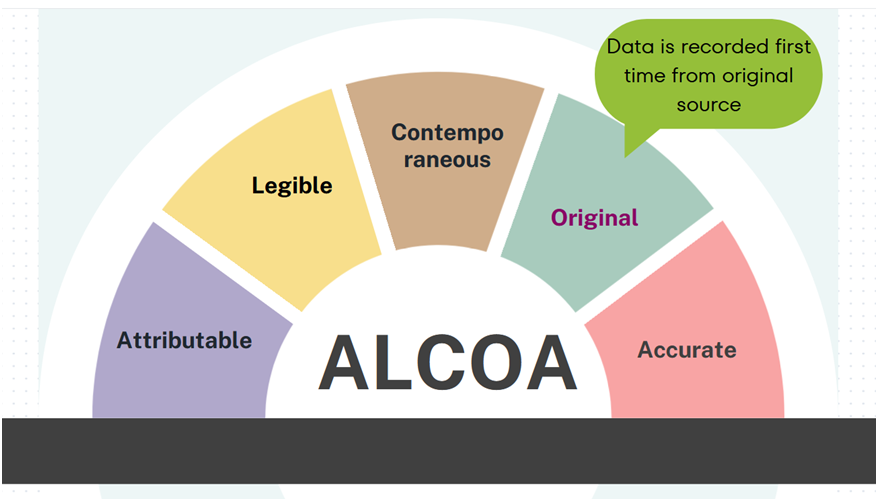
For example:
Ensure validation test results are recorded on the approved protocol. Recording results in a notebook for transcription later can introduce errors.
If original data is hand written and needs to be stored electronically, ensure a “true copy” is generated, the copy is verified for completeness and then migrated into the electronic system.
Accurate: For data and records to be accurate, they should be free from errors, complete, truthful and reflective of the observation. Editing should not be performed without documenting and annotating the amendments.
For example:
Use a witness check for critical record collection to confirm accuracy of data.
Place controls/verification on manual data entry, for example, temperature results can only be entered within a predefined range of 0-100°C.
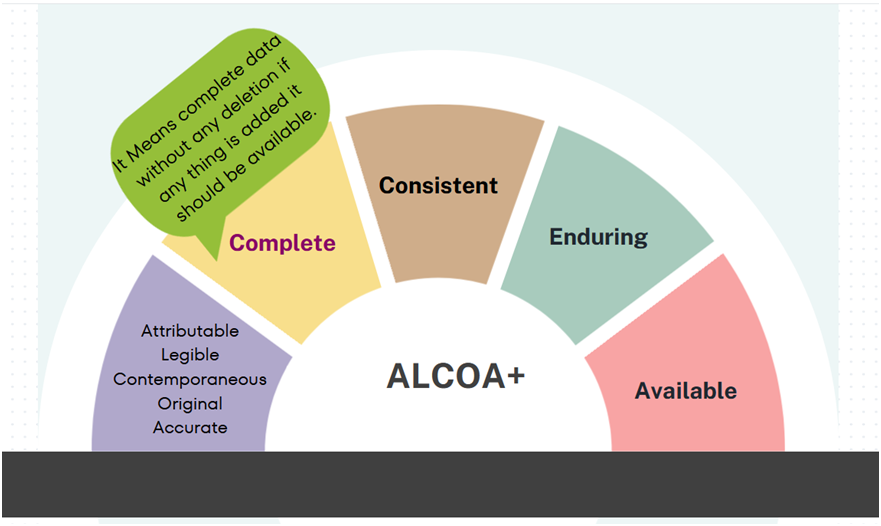
ALCOA+ (ALCOA Plus): A commonly used acronym for ‘Attributable, Legible, Contemporaneous, Original and Accurate’ which puts additional emphasis on the attributes being ‘Complete, Consistent, Enduring and Available’– qualities which are implicit in the basic ALCOA principles.
Complete: When data is complete in nature, it means there is no deletion that has taken place from the date of documenting. This include any changes that have been made during the life of the data.
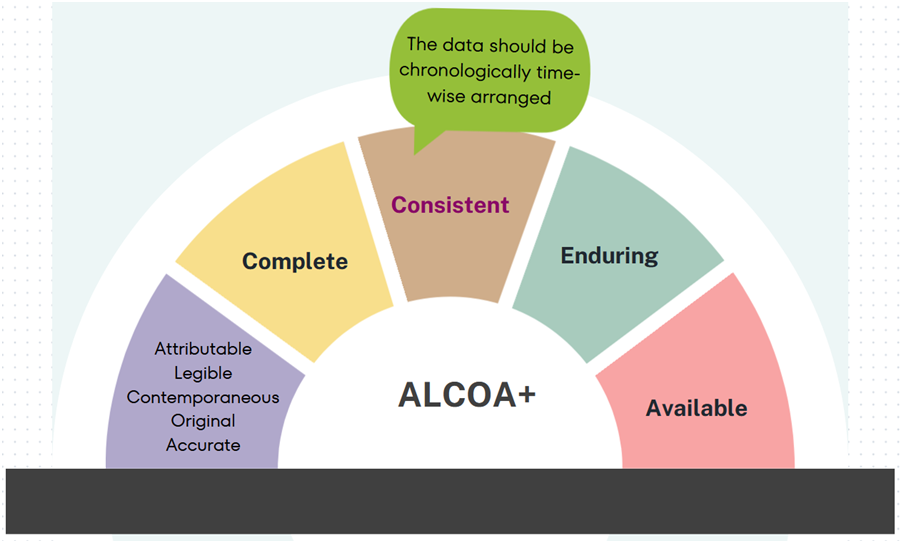
Consistent: The data should be chronologically arranged, with time stamps included for any addition to the original data. Consistency should be ensured by applying various audits over the life of the data.
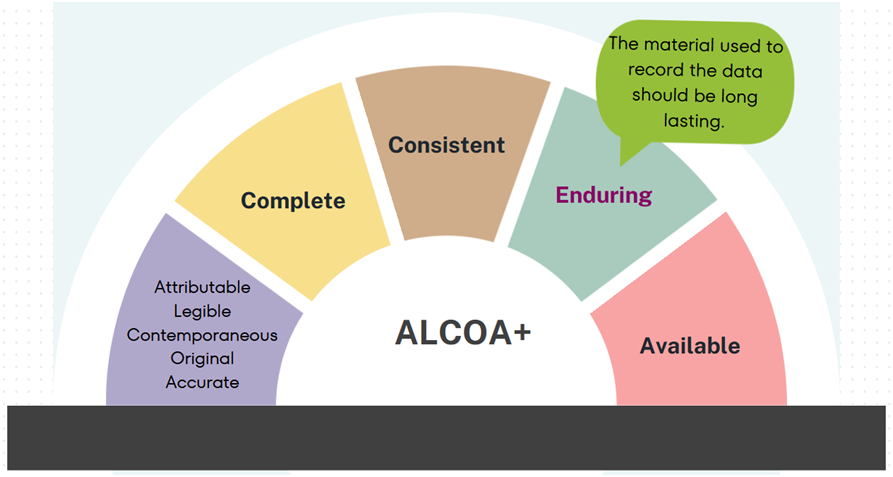
Enduring: The material used to record the data should be in manner which will last a long duration of time without losing the readability.
Available: Data should be accessible whenever needed, over the life of the data. Availability ensures the data meets it’s use, since can be applied when the need arises.
Data: The quantities, characters, or symbol on which operation are performed by a computer, which may be stored and transmitted in the form of electrical signals and recorded on magnetic, optical, or mechanical recording media or factual information(such as measurement or statistics) used as a basis for reasoning, discussion, or Calculation.
Controlled Copy: Controlled documents shall be posted server for read-only access or the hard copy shall be stamped “CONTROLLED COPY” in blue. Controlled copy shall be subject to automatic update when a new revision is released. It is for reference only.
Click the link to download word file copy of this document: https://pharmaguidehub.com/product/gxp-practices-in-pharmaceuticals/
Original/Master Copy: The original hard copy of the document that shall be approved and signed by authorized personnel.
- PRECAUTION:
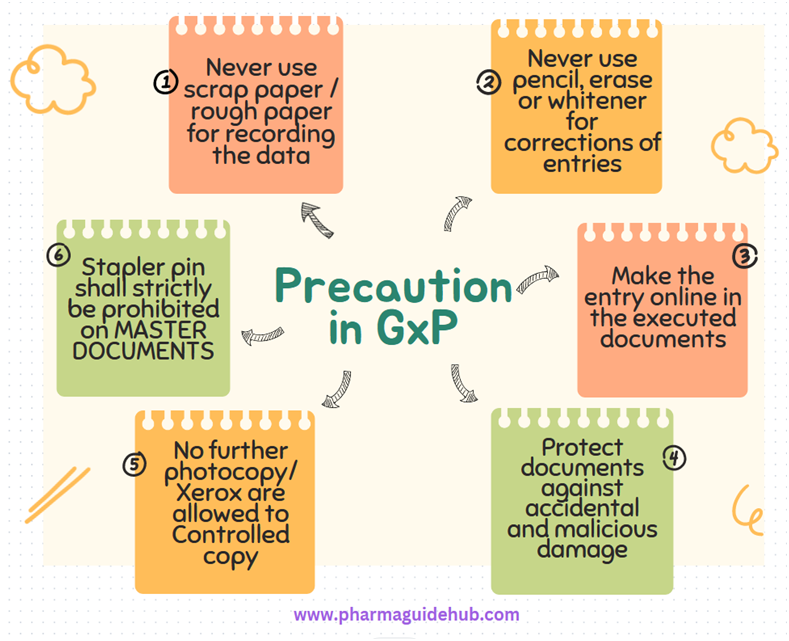
- Never use scrap paper / rough paper for recording the data. This means, all original observation / information shall always be entered directly the approved applicable format.Never use pencil, erase or whitener for corrections of entries.Make the entry online in the executed documents.Protect documents against accidental and malicious damage. Store documentation and associated information in a safe and secure manner.No further photocopy/Xerox are allowed to Controlled copy (if found shall be treated as Uncontrolled copy/copies).Stapler pin shall strictly be prohibited on MASTER DOCUMENTS, preserved in PVC folder/sheets.
- General Requirements for Documentation Practice
- All GxP documents shall be accurate, contemporaneous, legible and permanent, truthful and complete, readily retrievable and traceable.
- Data integrity shall be given utmost importance in GxP documentation as per SOP.
- All GxP activities shall be carried out with valid, correct and current effective versions of instruction documents and recording formats as per SOP on Document(s) and Data Control.
- Movement of any document (Manuals, QMS file etc.) from QA to other section and department shall be recorded.
- All controlled documents shall have a unique identification number and a revision number. The instruction source and unique identifier shall be documented in the respective record.
- For cases where Laboratory Notebooks, etc., are used during Development activities and where instructions are written manually in addition to recording experimental data, the Laboratory Notebook, though being an instruction document, may be excluded from this rule of requiring a version number; only an identification number needs to be provided.
- All documents/records shall have page numbers (preferred format: “Page X of Y”).
- All Master Documents shall be typed or preprinted. Hand written documents shall not be used as Master Documents.
- All Instruction documents shall have the effective date printed or stamped on them.
- Formats for date and time shall be described as given below:
- Date shall be written in one of the following formats:
Click the link to download word file copy of this document: https://pharmaguidehub.com/product/gxp-practices-in-pharmaceuticals/
DD MMM YYYY (For e.g. 11 MAY 2024 wherever stamp shall be used)
DD/MM/YYYY (For e.g. 11/05/2024 or 11/05/24 wherever hand written or reprinted).
Where,
“DD” is stand for the current date.
“MMM” is stand for first three letter of the current month mention in below table-01.“YYYY” is stand for current year.
“MM” is stand for the current month in numeric digit.
Table-1
| S.No. | MMM Indicates Alphabetic Months | MM Indicates two-digit numeric months |
| 1 | JAN indicate JANUARY | 01 |
| 2 | FEB indicate FEBRUARY | 02 |
| 3 | MAR indicate MARCH | 03 |
| 4 | APR indicate APRIL | 04 |
| 5 | MAY indicate MAY | 05 |
| 6 | JUN indicate JUNE | 06 |
| 7 | JUL indicate JULY | 07 |
| 8 | AUG indicate AUGUST | 08 |
| 9 | SEP indicate SEPTEMBER | 09 |
| 10 | OCT indicate OCTOBER | 10 |
| 11 | NOV indicate NOVEMBER | 11 |
| 12 | DEC indicate DECEMBER | 12 |
- All documentation of time and verification of time and date stamps shall be performed as per SOP on Data integrity and reliability.
- Time generated from all equipment and computers used for GxP activities shall be synchronized with the company clock provided in the area.
- Time shall be verified from the company clock provided in the area where the activity is being performed only.
- Time shall be recorded in the following formats:
For all the GMP records change the date after 24 hours cycle daily, i.e. after 23:59:60 or 00:00:00 in the night as per Indian Standard Time.
Time: Write time numerically in the form of HH:MM or HH:MM:SS, as applicable, in the document using 24 hours cycle daily. e.g.,
For 03:45 AM write 03:45 or 03:45:00
For 03:45 PM write 15:45 or 15:45:00
For 12:30 AM write 00:30 or 00:30:00
Time duration to be written as shown below:
Note:
- If for some process the observed time duration is 2 minutes and 30 seconds then it should be written as 2min 30 sec and not as 2:30min.
- In case time is printed from a machine or a computer, the time format of the machine or the computer shall be followed.
- Attachments shall be cross referenced to the parent document and the parent document shall be cross referenced to the attachments. In case of electronic records, all child or subsidiary records of a parent document shall have an indication of the relationship with the parent document.Data shall be recorded directly on approved and authorized formats only (e.g., batch production records, batch packaging records, laboratory notebooks, raw data sheets). Data for GxP documentation shall not be recorded on unauthorized documents (e.g. “Post It” sticky sheets, scrap papers, note pads).All GxP documents shall identify the significant steps that require checks by a second person while performing the activity (e.g. witnessing dispensing materials for batch production).Impermanent records like data printed on thermal paper, thin layer Chromatography (TLC) etc., shall be copied onto a permanent medium and the copies shall be attached to or stored along with, the original signed records.
- Handwritten Documents:
- Only indelible ball (preferably only blue colors) shall be used in GxP documents. The ink used shall be such that it can be photocopied.All departments shall use the permanent blue ball pen for execution of document. In document Issuance and other office work and for IPQA/IPQC activity black pen shall be used and same shall be used for GxP documents filling.Permanent BLUE indelible ball pen shall be used for signing of master documents for all departments. It is applicable for each and every department.QC supervisor shall use BLUE indelible ball pen for checking and approval only.The QA Person shall use the permanent Blue indelible ball pen to sign all GxP records, checking or approving the data.All entries shall be concise, legible, unambiguous and accurate.Handwritten records shall be signed or initiated and dated at the time the information is entered.All entries related to experiments being performed shall be done in chronological order.In the case of continuous pages of a notebook that are not being used to record data, continuity shall be denoted by recording the reference of the notebook number or page numbers at appropriate places. For example, if an experiment is recorded in a laboratory notebook on Page 25 and calculations are recorded on Page 35, a cross-reference linking the experiment with the calculations shall be recorded on both pages.
- The correct entry shall be written near to the strike out entry. The Person correcting the entry shall put the signature and date along with the corrected entry. It is preferable that the person who made the original entry shall strike through and shall make the correction. If this is not possible then correction shall be made in consultation with QA.
Example Suppose ‘Cleaned’ is recorded in place of ‘Ensured’ by mistake on 10/10/2024 and observed later during BMR review on 14/10/2024, it can be corrected at later date in the following way: Correct way for Correction on 14/10/2024: based on supporting data or investigation conclusion.
Cleaned Ensured. Sign/Date
If any observation / signature / date is to be repeated, the same shall be rewritten. Ditto (—-“—) marking or “as above” or “do” shall not be used.
Bracketing ( }) in any form shall not be used.
Click the link to download word file copy of this document: https://pharmaguidehub.com/product/gxp-practices-in-pharmaceuticals/
The following practices are strictly prohibited:
- Use of ditto marks (“) or down arrows ( ) to fill in repetitive entries.
- Use of pencil or any removable/water-soluble ball.
- Use of eraser or ball remover.
- Use of “white-out or correction fluid” to cover an entry and then write over it.
- Use of a stamp to replace manual dating, initials or signature on GMP documents, except in the case of validated electronic signature.
- When one option is to be selected from several text options, the correct option shall be preferably “encircled” or marked with “√”. Other option selection procedures may also be adopted; however, explanation of the use of these shall be provided in related procedures. An example is shown below:
- Working with Blank or Unused page/space:
Do not leave blank space in any cGxP record.
Blank spaces or pages shall have a single line through them with a signature and date and the reason for the page being blank (e.g. “Not Applicable”, “NA” or “N/A”).
- Signature Practices:
- Site shall maintain an updated master signature log wherein each employee involved in GxP activities shall provide their signatures and initials. The log shall be used for traceability of signatures for GxP records as per SOP.
- Signatures for all GxP activities shall always be accompanied by the relevant date wherever a separate date column has not been provided.
- Signatures indicate that the Signatory is responsible for the accuracy of data and information for the activity being signed for. The Signatory shall confirm the accuracy and completeness of information and data before signing.
- Persons preparing, reviewing or approving documents or persons recording, verifying or approving records shall sign and write the current date in the documents.
Note: Current date refers to the date when the document/record is signed.
For Example: A quality incident occurred on 05/10/2024 and the incident had an investigation conducted to identify the root cause. The investigation team completed the investigation and prepared a report. The investigation report was sent to one of the investigation team members, named “X” on 06/10/2024 for signature. Which date should be written along with X’s signature on the Investigation Report? – The date of the incident or the date when the Investigation Report was being signed?
Analysis of the Example:
Signature in the Investigation Report indicates that “X” has been part of the team investigating the incident and confirms that the investigation proceeded as per established procedures. X’s signature also denotes that said Investigation Report confirms the findings of the investigation and is truthful, accurate and complete.
Such confirmations are completed only once after the entire investigation has been completed and hence, the signature should bear the date on which the report is signed.
Importantly, back-dating is not allowed under any circumstances. “X” signing the Investigation Report on the date of the incident amounts to back-dating.
Hence, if “X” is signing the Investigation Report on 06/10/2024, he/she needs to put the same date along with his/her signature.
- All document signatories shall be adequately trained for the activity performed by them.
- For each activity/document (as applicable), each person shall sign (with current date) either as a Doer or a Verifier (also called “Checker”) or a Reviewer or an Approver. One person shall not sign for multiple roles for the same activity or entry (e.g. a doer cannot be the “Verifier”/ “Reviewer”/”Approver” for the same activity or entry recorded).
- No employee is authorized to sign for an activity performed by another employee.
- A clear meaning of each signature shall be provided (e.g. “Performed By”/” Verified By”/” Reviewed By”/” Approved By”).
- Definitions/Significance of Signatures:
Prepared By / performed By / Written By / Analyzed By (also called as “Doer”): The signature of the person actually carrying out the operation, test, inspection, calculation or other actions.
Entries in the documents/records along with Signature and Date shall be made at the time when the activity is performed (contemporaneously).
The signature of the “Doer” denotes that the “Doer” has performed the activity and confirms the authenticity of the data as that of the activity performed.
The Doer shall also check the result for its compliance against the specified limits/acceptance criteria and is expected to inform the respective Supervisor/HOD in case the results do not comply.
Verified By/Checked By: The signature of the person responsible for witnessing or conducting an independent check to ensure the operation, test, inspection, calculation or other actions followed required instructions and procedures and verifies the entries made by the Doer.
The “Verifier”/” Checker” shall record and sign concurrently for the ongoing activity being checked.
The signature of the “Verifier” denotes that the Verifier has confirmed that the entries are made correctly and are complying with predefined specifications/acceptance criteria.
Click the link to download word file copy of this document: https://pharmaguidehub.com/product/gxp-practices-in-pharmaceuticals/
Reviewed By: The signature of the person responsible for examining the documentation and certifying that the document/record was prepared / filled appropriately and in compliance with requirements.
The “Reviewer” shall review and sign (with date) for the activity/document/record being reviewed; the reviewer may or may not be present when the activity is being performed.
The “Reviewer” shall review completeness of the document/record and conformance of results recorded during the activity to established process parameters, limits and other applicable standards that define requirements of the activity being performed.
The signature of the “Reviewer” denotes that the document/record has been examined, all requirements have been fulfilled and the document/record demonstrates that the process was followed in accordance with the instructions provided.
Approved By: The signature of the person accepting the document/record for conformity to requirements.
The “Approver” shall review and sign (with date) for the activity/documents/record being approved; the Approver may or may not be present when the activity is being performed.
The Approver shall review conformance of results recorded during the activity to established process parameters, limits and other applicable standards that define requirements of the activity being performed.
The Signature of the “Approver” denotes that the document/record demonstrates that the process was followed in accordance with the instructions provided and is approved for conformity with requirements.
Authorized By: The signature of the person responsible for providing official permission or approval to another individual to perform a particular task.
Designee:
Reviewers/Approvers may delegate authority to another suitably qualified person to review/approve records, as applicable. The designee shall be qualified to perform the delegated task based upon relative job position, training, experience and subject matter expertise. Though a designee may perform the delegated task (of reviewing/approving, as applicable), final accountability of the activity performed by the designee shall reside with the person delegating the task. Supervisors of a signatory and/or members of the same department at equivalent or higher titles may function as designees without prior delegation of authority.
- The following signature practices are strictly prohibited:
- Pre-dating or post-dating (back dating) either documents or corrections.
- Pre-dating is completing an activity and then signing/dating that the activity was performed at a later time/date.
Example of Pre-Dating: An equipment is due for cleaning on 05/10/2024. The maintenance operator responsible for this activity has cleaned the tank on 01/10/2024; however, since this activity was scheduled for completion on 05/10/2024, the operator signs for completion of this activity with a date of 05/10/2024. This is called Pre-Dating, that is, signing for an activity in advance (i.e. with a non-current date).
- Back-Dating is completing an activity and then signing/dating that the activity was performed at an earlier time/date.
Example of Back-Dating: An equipment is due for cleaning on 05/10/2024. The maintenance operator responsible for this activity could not perform the cleaning activity on 05/10/2024, but performed the activity on 06/10/2024. However, he signs for completion of this activity with a date of 05/10/2024. This is called Back-Dating, that is, signing for an activity with a back-date (i.e. with a non-current date).
Click the link to download word file copy of this document: https://pharmaguidehub.com/product/gxp-practices-in-pharmaceuticals/
- Signing someone else’s name unless the signer is a Designee and it is clearly notated that the Designee has signed on behalf of the person, e.g., if Ram Kumar is a designated signer for Ajay Kumar, then Ram Kumar would sign as, “Ram Kumar for Ajay Kumar.
- Signing documents/records that are known to be containing information that is inaccurate, false, misleading and/or untruthful.
- Correction of Errors/Handling of Missed entries:
- DON’Ts: Entries in documents/records shall not be cancelled, erased obliterated or otherwise rendered illegible by using correction fluid/tape overwriting or crossing out with multiple strokes.
Example: Overwriting / Obliterating/Crossing out This is an unacceptable correction.
- DOs: When a correction is necessary, the erroneous/wrong entry shall be crossed out with a single horizontal line such that it shall not obscure the original entry. A brief reason for the correction shall be noted as to why the change was made and the correction shall be signed and dated.
Example: This is an acceptable correction as the original information must still be legible after the correction is made.
- When the reason for change requires lengthy explanation, it shall be clearly stated and shall be justified by supporting rationale. The reason may be in the form of a memorandum that is referenced in and attached to the original record. If this change affects the outcome of data, an Investigation shall be initiated and, post-investigation, correction of the error shall be made and the change shall be countersigned by a supervisor. Supporting documents shall be attached, if required.
- If there is insufficient space to enter a remark, then an annotation mark shall be placed near the incorrect entry and explained on the same page along with signature and date.
- Attempts to cover up mistakes are serious data integrity concerns and are strictly prohibited at all levels.
- The following conditions that may occur during correction of errors/completion of missed entries shall require evaluation as per current SOP of Deviation/Incident.
- The employee who made the error/person who missed recording data is not available in the organization.
- The information for an error/missed entry cannot be traced or determined.
- Errors/Missed Entries identified after a document/record has been approved/closed by QA.
- Requirement for correction of errors, including transcription/typographical errors related to data /missed entries in documents/records have already been submitted to Regulatory Agencies.
- Errors identified in approved documents while the activity is being performed.
- The following general rules shall be followed for correction of errors/handling of missed entries:
- All error corrections/filling of missed entries shall be done by the document “Doer”, irrespective of the time/date at which the error was noticed.
- If a worker (the “Doer”) made an error/missed an entry and they are no longer available due to reasons, such as leaving the organization or taking a leave for an extended period, such matters shall be escalated to the Department Head and an investigation shall be initiated.
- Based on the impact assessment and investigation outcome, another employee may be authorized to correct the error/fill in the missed entry as part of the corrective action. The employee shall provide an adequate justification and mention the name of the doer while performing the correction.
- Not all missed entries can be filled (corrected); if the information for filling in the missing data cannot be traced or determined, the Functional Manager or designee and QA Manager shall be informed immediately and shall take steps for further actions (including a decision for not filling in the missing entry) and provide explanations, which shall be recorded. The corrections/ explanations shall be countersigned by the QA Manager or designee. An investigation shall be completed and used by QA to determine the disposition of impacted product.
- The following general rules shall be followed for correction of errors/handling of missed entries:
Example of a batch record below shows discrepancy in mixing time of the blend:
Analysis of the example:
The Manufacturing Instruction (MI) lists that mixing time for the blend is not less than 15 minutes. In the batch record, Start time is 22:38, Stop time is 22:43; hence, the actual mixing time is five minutes instead of 15 minutes.
There is no supporting printout to support the actual blending time and mixing time is a very critical parameter of the manufacturing operation. Hence, this error cannot be corrected and the status of the batch is in jeopardy. Investigation and impact analysis shall be carried out and results used to justify the final batch disposition.
- Errors / Missed Entries identified at the time of verification / review / approval of a document / record may be managed at the level of verifier/reviewer/approver, as applicable; that is, the doer may correct the erroneous entry / fill in the missed entry and mark it as “Error Corrected”/”Late Entry” (as applicable) and sign (with current date) in the presence of the Verifier / Reviewer / Approver, as applicable.
- Errors / Missed Entries identified after a document has been approved / closed by QA shall be corrected / filled in (as applicable) by the doer only in the presence of QA and QA shall counter-sign near the correction.
- Typographical Errors/Missed Entries observed in “approved” documents during activity, shall be corrected / filled in (as applicable) on the respective page by the concerned supervisor, including signature and date and shall be verified by the QA Manager/designee.
- In case of a Transcription Error/Typographical Error/Missed Entry observed in a Summary/Certificate of Analysis (COA)/any other secondary compiled data, which is already submitted to Regulatory Agencies, Regulatory Affairs (RA) shall be notified of the incident and, if required, the responsible person shall submit a corrected summary identifying the cause for the revisions that shall be verified by QA and forwarded to RA for submission to regulatory agencies.
- If while stamping the effective date on documents or during retrospective review, it is discovered that an incorrect stamp was used, then the following procedure shall be used:
- The person identifying the error shall notify QA.
- The error shall be corrected by putting the correct stamp imprint adjacent to the incorrect one.
- The incorrect stamp imprint shall be struck off by “Doer” with single horizontal line in a manner that it shall be readable and not obscured.
- The “Doer” shall sign with date near the crossed out incorrect stamp imprint providing a rationale /justification; this activity shall be verified and signed (with date) by QA.
- Analytical/Manufacturing Documentation:
- Production officer and QC Analysts shall record actual results obtained at the time of performing an activity, without bias or prejudice.
- Only validated Excel spreadsheets shall be used for calculations wherever such Excel spreadsheets are not available, calculations shall be re-verified with qualified calculators.
- During the manufacturing process, sequential steps listed in the MI shall be directly recorded in the batch records as soon as the activity is performed.
- In case of laboratory analysis, step-by-step details of the testing procedure, dilutions, critical test parameters and date/time of activity, as required by the Standard Test Procedure (STP), shall be documented concurrently in Analytical template/worksheet/Laboratory notebooks or using controlled forms.
- A description of the sample received for testing with identification of source, quantity, lot number or other distinctive code, date sample was taken and date sample was received for testing shall be documented in the sample notebook or equivalent.
- All weight slips, printouts, chromatograms, spectrum, records of analysis, etc., shall be signed with date by the person performing the activity immediately after completing the activity and included with the respective record.
- If raw data prints/slips are too small to be included with the records (e.g., Batch Production Record (BPR), Record of Analysis, Qualification Documents), they shall be affixed on the specified stationary/space designated for them. These affixed printouts shall be cross-referenced to the parent documents and shall be enclosed with the parent record.
- All invalidated/disregarded chromatograms and other cGxP documents shall have supporting justification written by the Analyst performing the activity, be signed/dated and approved by relevant stakeholders.
- Wherever data/information for an activity must be captured electronically by an automated system (e.g. PLC, Data logger), the parent document shall contain instructions to attach copies of such printouts. All elements needed to associate the electronic records with the analysis and/or study shall be fully documented. These printouts shall be signed and dated. In such cases, the signature represents that the person performing the activity has verified that the printout is accurate and a complete reproduction of data/information taken from the electronic system.
- Site shall implement a “rounding rules” procedure for the standardized management of numerical values during manufacturing/ analysis as per respective SOP.
- Readings or values that are to be recorded from digital electronic displays shall be transcribed as they appear from the system to documents.
- Analytical/Manufacturing Documentation:
Example: If a balance is displaying 23.230 kg, then the value shall be recorded as 23.230 kg and not as 23.23 kg.
Click the link to download word file copy of this document: https://pharmaguidehub.com/product/gxp-practices-in-pharmaceuticals/
- In case a sample has been analyzed by two or more Analysts for different tests, each Analyst shall complete the test and related documentation for respective tests and sign (with date) his or her part. After ensuring the completion of all tests required per specification, including those sent to the contract laboratory for certain tests, the COA shall be prepared.
- Entries like “Complies/Does not comply” only allowed for the binary observations but the binary observation shall be specific .e.g. Limit test shall mention the observation noticed and TLC shall mention the comparison with the spot.
- Concordance of another analyst shall be taken for observations of subjective tests. e.g. limit tests, TLC plates etc. Both the analyst shall initialize and sign the observation.
- Printouts from the instruments relevant to the analysis shall be retained and no such document shall be discarded even if they are not of use in the calculation.
- Rounding – off of values:
- The rounding off values is applicable only to the calculations and not to the observed readings. Limits which are fixed numbers shall not be rounded off.
- Rounding – off of values:
For Example: If limit is 2 – 8 °C, then observed value 8.2 cannot be rounded off.
- When rounding off of any value is required, follow the below procedure.
- If last digit is equals to or greater than 5, it is eliminated and the preceding digit is increased by one.
- If last digit is smaller than 5, it is eliminated and the preceding digit remains unchanged.
- For examples see the below Table-02:
| Illustration of Rounding of Numerical Values for comparison with Requirements | ||
| Requirements | Unrounded Value | Rounded Value |
| Product Yield Limit (98.0% to 99.5%) | 98.926 % | 98.93 % |
| 98.124 % | 98.12 % | |
| 99.655 % | 99.66 % | |
- Assigning Due Date:
- For assigning Due Date in all GMP records, calculate due date as per frequency for that particular activity from the day on which that activity is performed.
- Status of the activity can be valid up to the due date.
For example, consider the case of assigning due date for re-cleaning of prefilters for which cleaning frequency is 15 days. Suppose cleaning of pre-filter is done on 01/05/2024, since frequency of prefilters is 15 days, due date for re-cleaning of prefilters can be assigned 15/05/2024 and cleaning can be considered valid up to 15/05/2024.
- Record all information in legible handwriting in all GMP records.
- Some typefaces have characters that are easily misread and create confusion. Few characters are listed below, which is not limited to this:
| 3 | 5 |
| 3 | 8 |
| 5 | 8 |
| S | 5 |
| 4 | 9 |
| 1 | I |
| 1 | 9 |
| 1 | 7 |
| 7 | 9 |
| c | e |
| f | P |
| 8 | g |
| 9 | g |
Requirements (GENERAL)
GxP documents shall be provide a clear, accurate history of an activity or event.
Procedures shall require that all entries in GxP documentation be permanent, legible, accurate, prompt, clear, consistent, complete, direct and truthful.
Only current, approved versions of SOPs or forms shall be used to perform cGxP activities. Systems shall be in place to prevent use of superseded documents.
Uncontrolled documents shall not be used in GMP areas.
Facsimile (Fax) can be used for GMP activities, but must be signed/dated to do so. In addition, the Fax shall be signed / dated by the recipient upon receipt.
Email from non-validated or unsecure systems should not be used as the primary document where a hardcopy is required. When there is no alternative, the responsible person should print out the email, sign and date as being received. Email may be used to confirm receipt of GMP documents in accordance with the requirements of this section. Documents in PDF (or other image formats) sent as an attachment to an email can be treated as GMP similar to a Fax.
Original records shall be kept in a secure location with controlled access.
Original records shall be stored with the batch documentation and archived by the respective documentation cell.
Copies of master documents / raw data shall not be allowed unless justified. When copies of master documents / raw data are required, for example during a regulatory inspection, they should be clearly identified as an “UNCONTROLLED COPY” or “CONFIDENTIAL”.
Use of scrap paper, post-it notes or loose (unbound) paper in GMP areas to record data is not permitted.
When printouts are made that require verification, e.g. proof of weight printout) and are not automatically verified by a validated electronic system, they shall signed/dated manually by personnel performing the work (Doer) and the person verifying the work, as required.
Procedures shall require that batch records include identification of the persons performing and directly supervising or checking each significant step in the operation.
Reviews to ensure documentation is complete and accurate shall be performed by a qualified individual who did not perform the task.Procedures shall require that reference to other GMP documents/records (i.e. deviation, discrepancy, CAPA or investigation numbers) shall be documented in the appropriate step of the document or in the comment section of the document.
The following elements shall be included, as applicable, when documenting a comment or event on a GMP document/record:
· What happened?
· When it happened (time)
· Where it happened
· Personnel involved
Requirements (Applicable For Laboratory Records):
All data generated within the laboratory shall be recorded. The intent/objective of the testing shall be recorded prior to the initiation of data acquisition. The requirements of the testing shall be covered by a specification, validated/qualified method, protocol or investigation.
Laboratory records shall meet the regulatory requirements. The requirements herein are applicable to both hardcopy and electronic records.
Clearly Written Documentation – All documents shall be accurate and recorded in a manner that prevents errors and ensure consistency. Sufficient space shall be provided for entries. If multiple documents or records are used in parallel for documentation, then each shall reference the other and be traceable by formal documentation numbers or record identification.
Notebook/Worksheets – Site SOPs shall describe the types of notebooks/worksheets that may be used to record data. These notebooks/worksheets shall be controlled and designed in such a fashion that pages cannot be removed or replaced.
Pagination – Unbound documents shall have page numbers, such as page XX of YY, to indicate the total number of pages in the document.
Laboratory records: shall include complete data derived for all tests necessary to assure compliance with established specifications and requirements, including examinations and assays.
Sample: A description of the sample received for testing with identification of source, quantity, lot number or other distinctive code, date sample was taken and date sample was received for testing.
Date/time: Where required, the date/time of activity shall be documented accurately.
Electronic Records:
Electronic records shall include elements for control of the process including but not limited to: implementation of an overarching data governance system, ensuring data integrity arrangements, appropriate management of the data life cycle and use of an audit trail.
Systems shall be designed to assure compliance and data integrity including but not limited to:
· Restricting control/access to templates used for recording data.
· Implementing user access rights that prevent amendments to data or audit trail information.
· Ensuring access to raw data for staff performing data checking activities.
· Using automated data capture or printers labelled directly to the equipment.
· Locating printers in close proximity to associated activities.
· Using clocks for recording timed events.
· Having persons performing observed task countersign record whenever possible.
· Recording the execution of critical operations contemporaneously by the user in single electronic transactions not combined with other operations.
Audit Trail:· When electronic records are used to capture, process, report or store raw data the system design should ensure retention of full audit trails, showing all changes to the data, while retaining previous and original data.
· All changes made to data should be associated with the person making those changes, including a time stamp and reason for making the change.
Click the link to download word file copy of this document: https://pharmaguidehub.com/product/gxp-practices-in-pharmaceuticals/· Users shall not have the capability to disable the audit trail function.
· Audit trail review shall be included as part of the routine GMP data review/approval process and should be documented.
· QA should periodically review a sampling of relevant audit trails, including raw data and metadata, as part of the self-inspection procedures to ensure data governance compliance.
Test Method: A statement of each method used in the testing of the sample. The statement shall indicate the location of data that establish that the methods used in the testing of the sample meet proper standards of accuracy and reliability, as applied to the product tested. (If the method employed is in the current revision of the United States Pharmacopeia, National Formulary, AOAC INTERNATIONAL, book of Methods or in other recognized standard references or is detailed in an approved new drug application and the referenced method is not modified, a statement indicating the method and reference will suffice).
· The method source and unique identifiers (e.g. version, issue date, etc.) shall be documented.
· Complete records shall be maintained of any modification of an established method employed in testing. Such records shall include the reason for the modification and data to support the valid use and shall align with site-specific change control procedures.
Specification and/or acceptance criteria: The identification of specification and/or acceptance criteria associated with the analysis or study shall be fully identified (e.g. specification number, specification version number, specification name and effective date or protocol number, protocol version number, protocol name and approval date).
Standards, Reagents and Solutions: Complete records shall be maintained of all testing and standardization of laboratory reference standards, reagents, volumetric solutions and standard solutions.
· Name, source, grade, date of receipt, expiration date and lot or unique identifier shall be provided for each.
· Data on or cross-reference to, the preparation and testing of reference standards, reagents and standard solutions.
· For quantitative standards, the actual strength (property value or calculation value) shall be recorded.
· Weights: A statement of the weight or measure of samples, standards and reagents used for each test, where appropriate.
· Weights taken shall be supported by balance printouts detailing the balance used, weights measured, time and date.
· Weighs for individual dosage units tested for Content Uniformity and Dissolution testing can be captured, even though they are not required for calculations. These weights can be very useful supporting data, especially during laboratory investigations.
Printouts, Graphs, Charts and Spectra: A complete record of all data secured in the course of each test, including all printouts, graphs, charts and spectra from laboratory instrumentation, properly identified to show the specific component, drug product container, closure, in-process material or drug product and lot tested.
· Any printouts, graphs, charts or spectra shall be appropriately identified, signed and dated and properly retained and crossed referenced.
· Where practical, they shall be permanently affixed (with glue or tape) to the notebook or controlled data sheet and be marked such that it would be obvious if they become separated (e.g. marked on a border). Otherwise, all individual pages of a data set shall be maintained and secured together as a packet preventing the intentional or unintentional misplacement of the individual pages.
· Laboratory Instruments, Apparatus, Gauges and Recording Devices:
· Complete records shall be maintained of the periodic calibration of laboratory instruments, apparatus, gauges and recording devices.
· Records shall include validations, calibrations, maintenance and cleaning or repair operations.
· Instrument logs can be used to record the daily instrument performance verification check in addition to any instrument incident and unscheduled repairs.
· All pertinent instrument parameters need to be documented.
Calculations: A record of a calculation example and all calculation factors in connection with the test, including units of measure, conversion factors and equivalency factors shall be documented.
Results: There shall be a statement of the results of tests and how the results compare with the established specifications or acceptance criteria of identity, strength, quality and purity for the component, drug product container, closure, in-process material or drug product tested.
Signatures and date: The initials or signature of the person who performs each test and the date(s) the tests were performed.
· Each notebook/worksheet/template/form page shall be dated with a start date and signed and dated on completion of the page; or if not completed, at the end of the scheduled work day.
· The initials or signature of a second person and the review date showing that the original records have been reviewed for accuracy, completeness and compliance with established standards.
Electronic Records and Electronic Signatures:· Persons who use closed systems to create, modify, maintain or transmit electronic records shall employ procedures and controls designed to ensure the authenticity, integrity and, when appropriate, the confidentiality of electronic records and to ensure that the signer cannot readily repudiate the signed record as not genuine.
· Signature manifestation information should be subject to all controls required for electronic records and should include the following:
· Printed name of signer.
· Date and time signature was executed.
· Meaning associated with signature (e.g. review, approval, authorship).
Retention of Documents:
· It shall be clearly defined which record is related to each manufacturing activity and where this record is located. Secure controls must be in place to ensure the integrity of the record throughout the retention period and validated where appropriate.
· GMP documents are confidential and are the property of the company and all documents shall be returned to company archives for proper storage.
· Ensuring Proper Security and Storage of Documents, during Review Process. Precautions must be taken to prevent unauthorized persons from entering storage areas. In particular, storage area shall be clean, dry & free from fire hazard, accumulated waste and vermin and maintained within acceptable temperature, humidity & light exposure limits.
· Document storage area access shall be restricted to authorized personnel only.
· Retention period of all GxP documents as per site specified SOP.
· All GxP documents shall be retained for a pre-designated period. Following the expiration of the retention period, GxP documents shall be destroyed in a controlled manner as per SOP on Document(s) and Data Control.
REFERENCES:
Not Applicable
ANNEXURES:
Not Applicable
DISTRIBUTION:
· Control copy No. 1
:
Head Quality Assurance
· Control copy No. 2
:
Head Quality Control
· Control copy No. 3
:
Head Production
· Control copy No. 4
:
Head Purchase
· Control Copy No.5
:
Head Warehouse
· Control Copy No.6
Research and Development
· Control Copy No.7
Regulatory Affairs
· Master Copy
:
Quality Assurance Department
ABBREVIATIONS:
cGMP
:
Current Good Manufacturing Practices
GDP
:
Good Documentation Practice
PLC
:
Programmable logical controller
GxP
:
Good x Practices / good anything
REVISION HISTORY:
CHANGE HISTORY LOG
Revision No.
Details of Changes
Reason for Change
Effective Date
00
New SOP
Not Applicable
To be written manual
Frequently Asked Questions?
- What is GxP?
- GxP is a general term used in the pharmaceutical and healthcare industries to refer to various regulatory guidelines and practices, including Good Manufacturing Practice (GMP), Good Laboratory Practice (GLP), and Good Clinical Practice (GCP).
- What is the purpose of GxP regulations?
- GxP regulations are in place to ensure the quality, safety, and efficacy of pharmaceutical products. They provide a framework for manufacturing, testing, and distributing pharmaceuticals while maintaining compliance with regulatory standards.
- What is the difference between GMP, GLP, and GCP?
- GMP focuses on the manufacturing and quality control of pharmaceutical products, GLP pertains to the conduct of non-clinical laboratory studies, and GCP is related to the conduct of clinical trials.
- How does GxP impact pharmaceutical manufacturing?
- GxP ensures that pharmaceutical manufacturing processes are conducted in a controlled and consistent manner, minimizing the risk of contamination, errors, and deviations that could affect product quality.
- What is a deviation in GxP?
- A deviation is an unplanned divergence or departure from standard operating procedures. Managing deviations is crucial in GxP environments to ensure the identification, investigation, and resolution of issues that may impact product quality.
- How is data integrity maintained in GxP?
- Data integrity is maintained by implementing controls to ensure accuracy, reliability, and consistency of data throughout its lifecycle. This includes proper documentation, electronic record-keeping, and audit trails.
Click the link to download word file copy of this document: https://pharmaguidehub.com/product/gxp-practices-in-pharmaceuticals/
7. What is a validation process in GxP?
- Validation is the documented evidence that a system or process meets predefined requirements and specifications. It ensures that equipment, processes, and systems are capable of consistently producing results within established parameters.
8. How often should equipment be calibrated in a GxP environment?
- The frequency of equipment calibration depends on the type of equipment and its intended use. Typically, calibration is performed at regular intervals, and the schedule is defined in standard operating procedures.
9. What is the role of Quality Risk Management (QRM) in GxP?
- Quality Risk Management is a systematic process for assessing, controlling, and managing risks to product quality. It helps pharmaceutical companies identify and mitigate potential risks throughout the product lifecycle.
10. How are personnel trained in GxP compliance?
- Personnel are trained through a structured training program that covers GxP regulations, standard operating procedures, and job-specific tasks. Training records are maintained to demonstrate compliance with training requirements.
Click the link to download word file copy of this document: https://pharmaguidehub.com/product/gxp-practices-in-pharmaceuticals/

