
- OBJECTIVE:
The objective of this document is to define the approach for Hold Time Studies of Non-sterile formulations and will ensure compliance with National and International GMP.
- SCOPE:
This Policy is applicable to the hold time studies for non-sterile formulations manufactured at {Company Name} {Location}.
- RESPONSIBILITY:

- Quality Assurance: To prepare Protocol and report for hold time studies.
- QA Department Person:
- QA shall be responsible for collecting the hold time samples as per the hold time study Protocol/Datasheet and shall send duly filled “Sample request slip” to the QC department.
- QA shall be responsible for maintaining the hold time register and soft copy of the hold day’s record.
- QA shall be responsible for investigating in case of failure/discrepancies
- QC Department Person:
- QC shall be responsible for making entries of the sample as per the defined procedure and analyzing the sample as per the defined procedure.
- QC shall be responsible to share the report and investigate in case of failure/discrepancies.
Click the link to download word file copy of this document: https://pharmaguidehub.com/product/hold-time-studies-for-non-sterile-formulations-in-pharmaceutical/
- Production Department Person:
- Production shall be responsible to prepare the Test Requisition cum report and intimate to QA for sampling.
- Quality Head and Plant Head Responsible for:
- Quality Head and Plant Head shall be responsible for reviewing and approving the SOP.
- Responsible for the implementation of the defined system.
- ACCOUNTABILITY:
QA Head shall be Accountable for implementation of SOP.
- PROCEDURE:
DEFINITIONS: The definitions listed here apply in the context of this Policy Document and may or may not be applicable in all other usage.
- Acceptance criteria – The criteria a system/ product/ process must meet to successfully complete a test phase or to achieve delivery requirements.
- Bulk product– A pharmaceutical product that has completed all processing stages up to, but not including, final packaging.
- Hold time – The established time period for which dispensed raw materials, intermediates (in-process materials) and bulk dosage form awaiting final packing, may be held under specified conditions and will remain within the defined specifications. Intermediate (in-process materials): Partly processed product that must undergo further manufacturing steps before it becomes a bulk product.
- Dispensed Material: A set of Dispensed active & excipient raw materials as per the approved formula comes under dispensed material.
- Intermediate: A partly processed product that must undergo further manufacturing steps before it becomes a bulk product.
- Bulk Product: Any pharmaceutical product that has completed all processing stages up to, but not including final packaging.
- DOCUMENTATION REQUIREMENTS:
- Procedure for conducting hold time studies for non-sterile formulations.
- Protocol and report for hold time studies.
- POLICY:
- General Considerations:
- Hold time studies shall establish the time limits for holding the materials at different stages of production (i.e. dispensed raw materials, intermediates and bulk dosage form awaiting final packing) to ensure that the quality of the product does not produce results outside the acceptance criteria during the hold time.
- Hold times should normally to be determined prior to marketing of a product.
- Process flow charts shall be used to review the manufacturing process for identifying the critical stages of the process to evaluate the potential impact of storage with reference to environmental condition.
- Hold time studies shall be established for every product through an approved protocol.
- Bracketing approach shall be followed, in case of multiple strengths of the same product, hold time study shall be performed on either on the highest strength (if different strengths are dose proportional) or on both on the lowest and highest strengths of the product (if different strengths are not dose proportional).
- Hold time studies shall be performed for material, intermediate and bulk product on exhibit or commercial batches.
- A risk-based approach can be used to determine the appropriate number of batches considering the characteristics of the material and other relevant aspects.
- The product knowledge gained during the development studies shall be considered for the Hold time study requirements.
- The test parameters for the hold time study shall be defined based on the sensitivity of product. Some of the examples for various process stages, where applicable are, but not limited to, following parameters:
- POLICY:
Click the link to download word file copy of this document: https://pharmaguidehub.com/product/hold-time-studies-for-non-sterile-formulations-in-pharmaceutical/

- Intermediate, Blend: Description, LOD/Water, Assay, Related Substances and Microbial test.
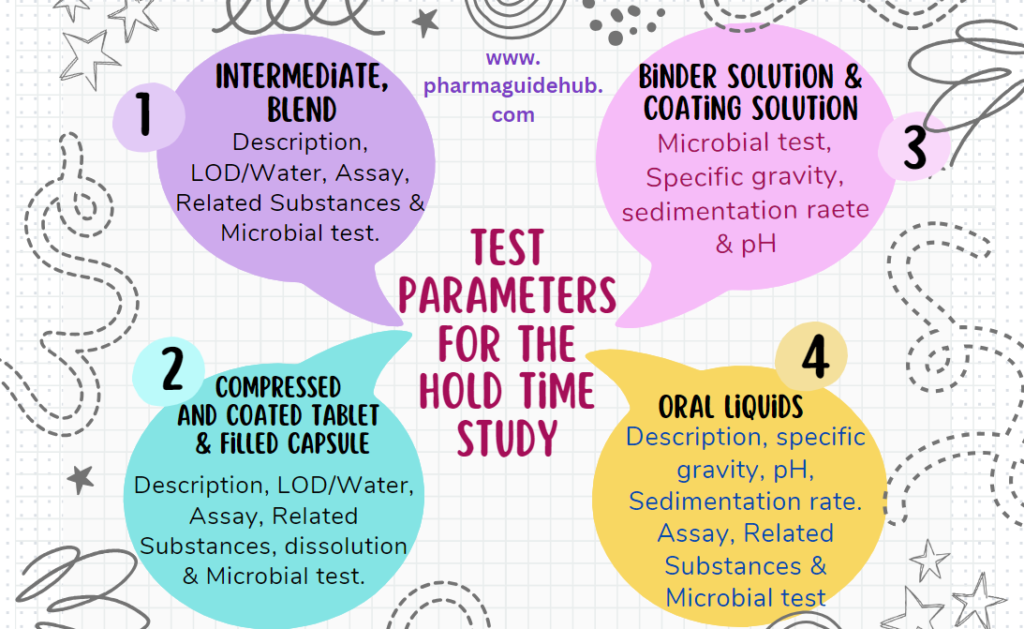
- Compressed and coated tablet filled capsule: Description, LOD/Water, Assay, Related Substances, dissolution and Microbial test.
- Binder solution and Coating solution: microbial test, pH.
- Oral Liquids/Suspension: Description, specific gravity, pH, Sedimentation rate. Assay, Related Substances and Microbial test.
- Batches subjected to hold time studies shall be considered for long-term stability.
- Hold Time Studies:
- Hold time studies shall be defined to ensure that dispensed materials, intermediate and bulk products if stored at recommended packaging and storage conditions, then it can be processed within the defined hold period.
- As an example, for oral tablets that are uncoated/coated, capsule, liquids and Dry Syrups the following stages (but not limited to) may be considered:
- Binder solution
- Wet mass
- Lubricated Blend
- Bulk oral liquid/suspension
- Coating Solution/dispersion
- Beads or pellets (Drug coating, Seal coating, Functional coating, Enteric coating etc)
- Core/Coated Tablets
- Filled capsules
- The study period and testing interval shall be determined based on the sensitivity of the product with environmental conditions prevailed in the manufacturing and storage areas. The possible maximum hold time during an actual production run shall also be considered.A representative sample of the batch of materials, intermediate or bulk product shall be collected from different locations as defined in a sampling diagram.The sample quantity for the hold time studies shall be determined based on the batch size, the intervals and the tests to be performed.Sample shall be stored in the original container or simulated container (constructed of the same material and using the same closure system). In case of reducing the size of container, for testing holding time, shall be justified.The headspace in the sample -holding container used for hold time studies shall be similar or higher than that of the actual headspace exist in the holding container used for manufacturing.The environmental conditions for hold time study sample storage shall be the same as those of the hold area/manufacture stage.Withdraw sample from the hold time study containers as per the time intervals defined in the respective hold time study protocol.The amount of samples required for particular time intervals shall be determined based on the tests to be performed.Samples shall be subjected for analysis (chemical and microbial) specified in the respective hold time study protocol.Acceptance criteria are typically more stringent than the product specification.If any finished/ semi-finished product crosses its defined hold time frequency then raise the QMS Document for justification, The semi-finished/Finished product shall be retested before proceeding to the next stage or the final decision shall be taken by the QA Head.Hold time sample shall be withdrawn in next working day in case of a holiday.
Click the link to download word file copy of this document: https://pharmaguidehub.com/product/hold-time-studies-for-non-sterile-formulations-in-pharmaceutical/
- Study time as per guideline – In-house -Frequency may vary as per protocol but not exceed frequency as per recommendation in WHO guidelines.
- Granulating fluid and coating suspension – Physical appearance, specific gravity, sedimentation, viscosity, pH, Microbial test – Initial, 12, 24, 36, 48, 60 & 72 hoursLiquid suspension & syrups – Physical appearance, specific gravity, sedimentation, viscosity, pH, Microbial test (For holding and manufacturing vessel) – Initial, after 24, 48 & 72 hours
Granular blend/Pellets – Description, assay, Loss on drying, Water content, blend uniformity, particle size, bulk/tapped density, Microbial test – Initial, 15th, 30th & 45th dayCore tablets- uncoated – Description, hardness, thickness, Friability, Average Weight, Disintegration, dissolution, related substances, uniformity of dosage unit, Assay, Microbial test – Initial, 30th, 45th, 60th & 90thday.Coated tablets – Description, appearance & Visual examination, hardness, thickness, Friability, Average Weight, Disintegration, dissolution, related substances, Assay, Microbial test – Initial, 30th, 45th, 60th & 90thday.Hold granulating fluid/coating suspension for 72 hours (samples send to QC at initial, end of 24 & 48 hours).Hold blend for 45 days (sample sent to QC at Initial, 15th 30th & 45thday)Hold core tablet for 90 days (sample sent to QC at Initial, 30th, 45th, 60th & 90th)Hold Coated tablets for 90 days (sample sent to QC at Initial, 30th, 45th, 60th & 90thday).
- Granulating fluid and coating suspension – Physical appearance, specific gravity, sedimentation, viscosity, pH, Microbial test – Initial, 12, 24, 36, 48, 60 & 72 hoursLiquid suspension & syrups – Physical appearance, specific gravity, sedimentation, viscosity, pH, Microbial test (For holding and manufacturing vessel) – Initial, after 24, 48 & 72 hours
- Study time as per guideline – In-house -Frequency may vary as per protocol but not exceed frequency as per recommendation in WHO guidelines.

- Hold time samples (Granulating fluid, Blends, Core tablets, Coated tablets & Coating Suspensions) handling during tablet manufacturing:
- Raw Materials:
- QA person shall collect the required quantity of samples of starting materials as per the protocol and store them in the HDPE container in quarantine.
- The entry of hold time samples collected should be made in the hold time sampling register.
- On completion of hold time, the ‘Test Requisition Cum Report’ should be filled by QA and sent to QC along with the sample.
- Granulating Fluid:
- QA person shall collect the required quantity of sample of granulating fluid (binder) as per the protocol and store it in the S.S. container in a controlled area.
- The entry of hold time samples collected should be made in the hold time sampling register.
- On completion of hold time, the ‘Test Requisition Cum Report’ should be filled by QA and sent to QC along with the sample.
- Blend Stage:
- QA person shall collect the required quantity of the sample of blend/granules/pellets as per the protocol or as specified in BMR/MPS and stored in the HDPE container in blend Quarantine.
- The entry of hold time samples collected should be made in the hold time sampling register.
- On completion of hold time, the ‘Test Requisition Cum Report’ should be filled by QA and sent to QC along with the sample.
- Compression Stage:
- QA person shall collect the required quantity of the sample of the core tablet as per the protocol and store it in the HDPE container in tablet Quarantine.
- The entry of hold time samples collected shall be made in the hold time sampling register.
- On completion of hold time, the ‘Test Requisition Cum Report’ shall be filled by QA and sent to QC along with the sample.
- Coating Stage:
- QA person shall collect the required quantity of the sample of the coated tablet as per the protocol and store it in the HDPE container in tablet Quarantine.
- The entry of hold time samples collected should be made in the hold time sampling register.
- On completion of hold time, the ‘Test Requisition Cum Report’ should be filled by QA and sent to QC along with the sample.
- Coating Suspension:
- QA person shall collect the required quantity of coating suspension as per the protocol and store it in the S.S. container in the tablet Quarantine.
- The entry of hold time samples collected should be made in the hold time sampling register.
- On completion of hold time, the ‘Test Requisition Cum Report’ should be filled by QA and sent to QC along with the sample.
- Documentation:
- Hold time studies shall be performed using approved protocols.
- The protocols may have the following content but are not limited to
- Documentation:
Click the link to download word file copy of this document: https://pharmaguidehub.com/product/hold-time-studies-for-non-sterile-formulations-in-pharmaceutical/
- Objective of the study
- Scope
- Responsibilities
- Procedure
- Description of material/product
- Sample quantities
- Sampling method and criteria
- Acceptance limits
- Frequency of sampling
- Sampling locations
- Storage conditions
- Type of container
- methods of analysis etc.
- Hold time study reports shall have the results of batch(s) studied, compilation of the non-conformances noticed during study, summary of results, conclusion and recommendation.
- Training Aspects: Personnel involved in the hold time study shall be adequately trained of the study protocols in all aspects including techniques related to sampling, sample handling and analysis.
- Hold time study Failure Considerations: Whenever a failure is observed during hold time study, Process nonconformance shall be initiated and an investigation shall be conducted. Root cause or probable cause shall be determined and an adequate control measure shall be put in place. In such cases, the decision for assignment of hold time period shall base on the conclusion from the investigation.
- Review and Revision: The risk assessment of changes to processes, equipment, storage conditions, starting or packaging materials shall include an assessment of whether further hold-time studies should be performed. Based on the satisfactory compliance of hold time study report recommendations, change control shall be raised to update the manufacturing documents.
- REFERENCES:
Not Applicable
- ANNEXURES:
Not Applicable
ENCLOSURES: SOP Training Record.
- DISTRIBUTION:
- Controlled Copy No. 01 : Head Quality Assurance
- Controlled Copy No. 02 : Head Production
- Controlled Copy No. 03 : Head QC
- Controlled Copy No. 04 : Head warehouse
- Master Copy : Quality Assurance Department
- ABBREVIATIONS:
| QA | : | Quality Assurance |
| No. | : | Number |
| SOP | : | Standard Operating Procedure |
| LOD | : | Loss on Drying |
- REVISION HISTORY:
CHANGE HISTORY LOG
| Revision No. | Details of Changes | Reason for Change | Effective Date |
| 00 | New SOP | Not Applicable | To Be Enter Manual |
Click the link to download word file copy of this document: https://pharmaguidehub.com/product/hold-time-studies-for-non-sterile-formulations-in-pharmaceutical/
Frequently Asked Questions ?
Q1: What is a hold time study in the pharmaceutical industry?
A1: A hold time study in the pharmaceutical industry refers to the systematic evaluation of the duration during which a pharmaceutical product can be safely stored or held before further processing or packaging. It is crucial for ensuring product stability, quality, and compliance with regulatory requirements.
Q2: Why is a hold time study necessary in pharmaceutical manufacturing?
A2: Hold time studies are essential to determine the maximum allowable time a pharmaceutical product can be held in a specific stage of the manufacturing process without compromising its quality, safety, or efficacy. These studies help establish appropriate procedures and guidelines to maintain product integrity.
Q3: What factors are considered in a hold time study?
A3: Factors considered in a hold time study include the nature of the pharmaceutical product, storage conditions, temperature, humidity, and the specific stage of the manufacturing process. Additionally, the study may assess the impact of hold times on the stability of the product and its compliance with regulatory standards.
Q4: How is a hold time study conducted in pharmaceutical manufacturing?
A4: A hold time study involves designing experiments to assess the impact of extended holding periods on the product. Samples are analyzed for various quality attributes at different time points to identify any degradation or changes. Data collected is then used to establish safe and effective hold times.
Q5: What are the regulatory implications of a hold time study?
A5: Regulatory agencies, such as the FDA, often require pharmaceutical manufacturers to conduct hold time studies as part of Good Manufacturing Practice (GMP) regulations. Compliance ensures that the product meets quality standards, and the study results are often submitted as part of regulatory documentation.
Q6: How does a successful hold time study benefit pharmaceutical companies?
A6: A successful hold time study helps pharmaceutical companies optimize their manufacturing processes by providing assurance that products can be safely held without compromising quality. This can lead to increased efficiency, reduced production costs, and compliance with regulatory requirements.
Q7: Can hold time studies vary between different pharmaceutical products?
A7: Yes, hold time studies are product-specific. The characteristics and formulations of different pharmaceutical products may react differently to extended holding periods. Therefore, each product requires a tailored hold time study to ensure its stability and quality.
Q8: How often should hold time studies be reviewed and updated?
A8: Hold time studies should be periodically reviewed and updated, especially when there are changes in the manufacturing process, formulation, or equipment. Regular reassessment ensures that hold times remain accurate and align with current regulatory standards and product specifications.
Click the link to download word file copy of this document: https://pharmaguidehub.com/product/hold-time-studies-for-non-sterile-formulations-in-pharmaceutical/


