
- PROCEDURE FOR THE HANDLING OF OOS:

- Laboratory Tests are categorized in to:
- Chemistry based tests
- Physical tests like description, appearance, colour, non-conformance with approved reference shade card / art-work etc.,
- Microbiology tests
- For tests with tiers or stages, such as Uniformity of Dosage Units, Disintegration, Dissolution, or Drug Release, the result is not considered out of specification until the acceptance criteria for the final tier or stage are not met.
- Note: This failure to meet the acceptance criteria may occur before all tiers or stages of testing are completed. For example, an immediate release product may produce an out of specification test result during stage 1 testing if a single unit has a result less than Q – 25%. In such cases, an OOS investigation is initiated, and the tiered/staged test is continued through all tiers/stages to gather data for the investigation.
Click the link for download word file copy of this document: https://pharmaguidehub.com/product/investigation-of-out-of-specification/
- Handling of OOS results of Chemistry based tests:
- The Out of specification investigation related to Chemistry based tests shall be carried out in two phases
- Phase-I: Laboratory investigation
- Phase-II: Full scale investigation (Manufacturing Investigation and extended laboratory investigation.)
- Phase I investigation shall be carried out to identify the Obvious/Assignable laboratory cause.
- Phase II full scale investigation shall be conducted when the Phase I investigation did not reveal an assignable laboratory cause.
- This also includes the initial manufacturing investigation to determine whether there was a possible manufacturing root cause.
- All standard, sample preparations, other solutions, reagents, and glassware must be retained until all data has been second person verified as being within the defined acceptance criteria.
- After completion of analysis, the analyst shall enter the data in logbooks.Upon observing of OOS result, analyst shall notify (intimate) the section in-charge immediately, (i.e., within the same day and same shift of day), for the potential need to initiate an investigation.
- QC personnel shall communicate to QA about OOS occurrence and obtain OOS Number and “Out of specification investigation summary report” (Format-I).
- If confirmed OOS for stability batch(es) and subjected batch(es) are distributed in US market, follow the procedure specified as per the SOP.
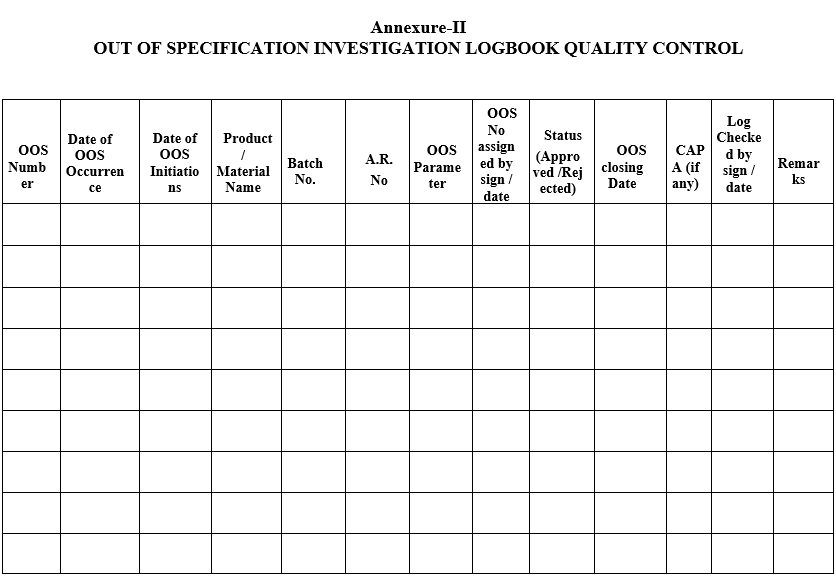
- QA executive shall register the OOS details in Out of specification investigation logbook.
- Refer Format-II for QC and Format-III for Stability.
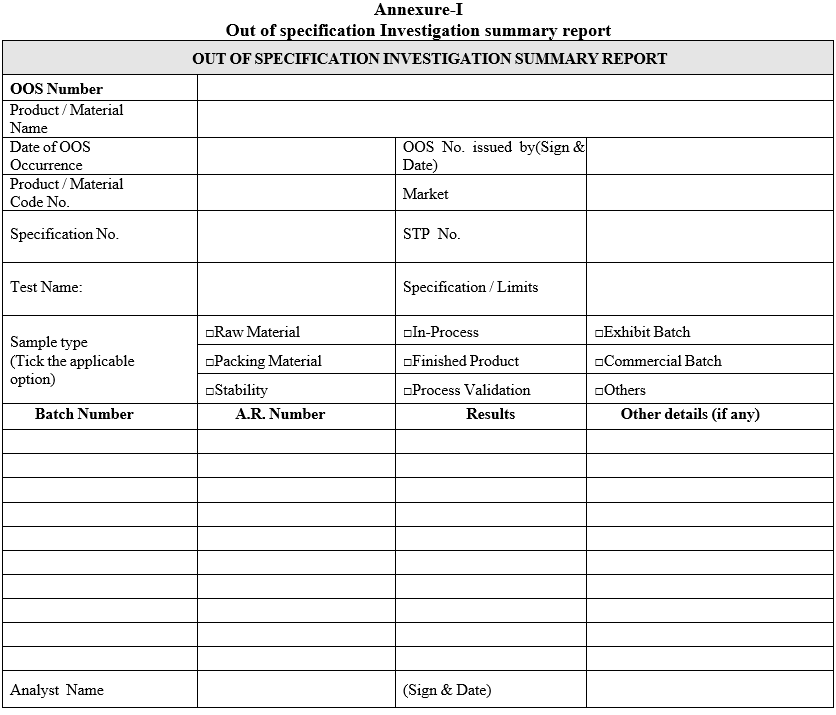
- “Out of specification investigation summary report” (Format-I) shall be issued for each occurrence for the affected batch(es) for each respective test.
- Out of Specification numbering shall be given as below.
- In case of Quality Control Department:
QC/OOS/YY/NNN
| QC | : | Quality control department |
| / | : | Slash |
| OOS | : | Out of specification |
| / | : | Slash |
| YY | : | Last two digits of current year |
| / | : | Slash |
| NNN | : | Sr. no. stating from 001,002 and so on. |
- In case of Stability Department:
ST/OOS/YY/NNN
| ST | : | Stability |
| / | : | Slash |
| OOS | : | Out of specification |
| / | : | Slash |
| YY | : | Last two digits of current year |
| / | : | Slash |
| NNN | : | Sr. no. stating from 001,002 and so on. |
- The investigation shall be carried out as per the flow chart.
- The Out of Specification Laboratory Investigation Report shall be prepared as per Format -V wherever required.
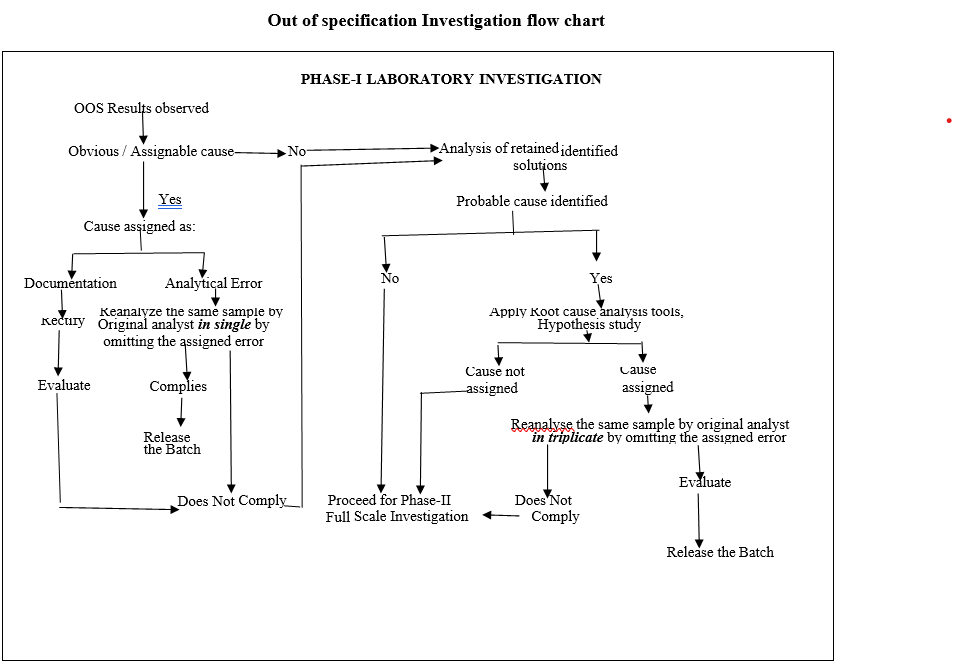
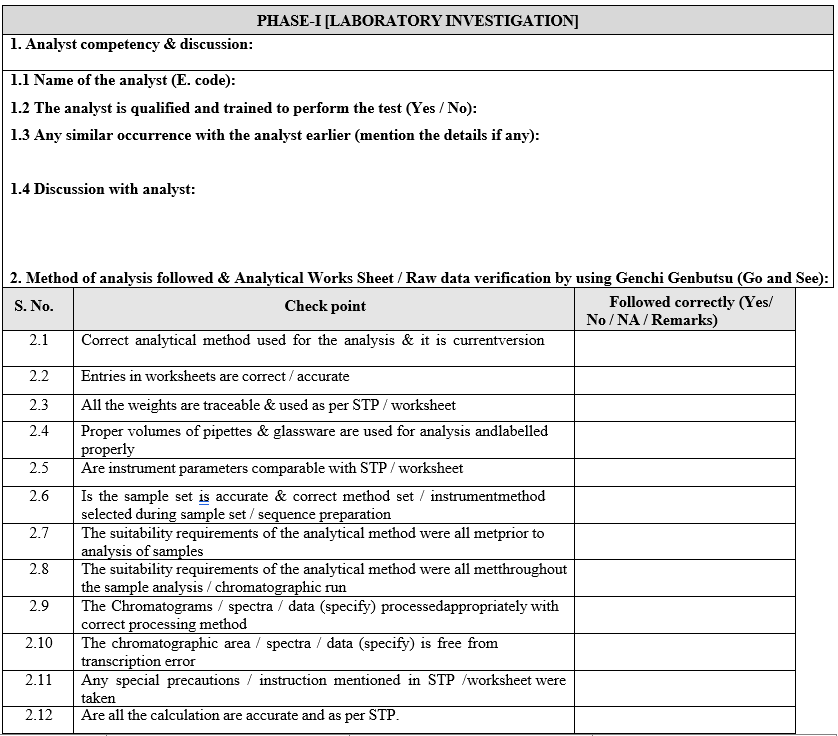
- Phase-I Laboratory Investigation:
- The Section In charge/designee shall initiate the investigation along with analyst and analytical QA executive/designee.
- The preliminary (i.e. obvious / assignable cause) investigation shall be carried out as given in Format-I (Out of specification Investigation summary report).
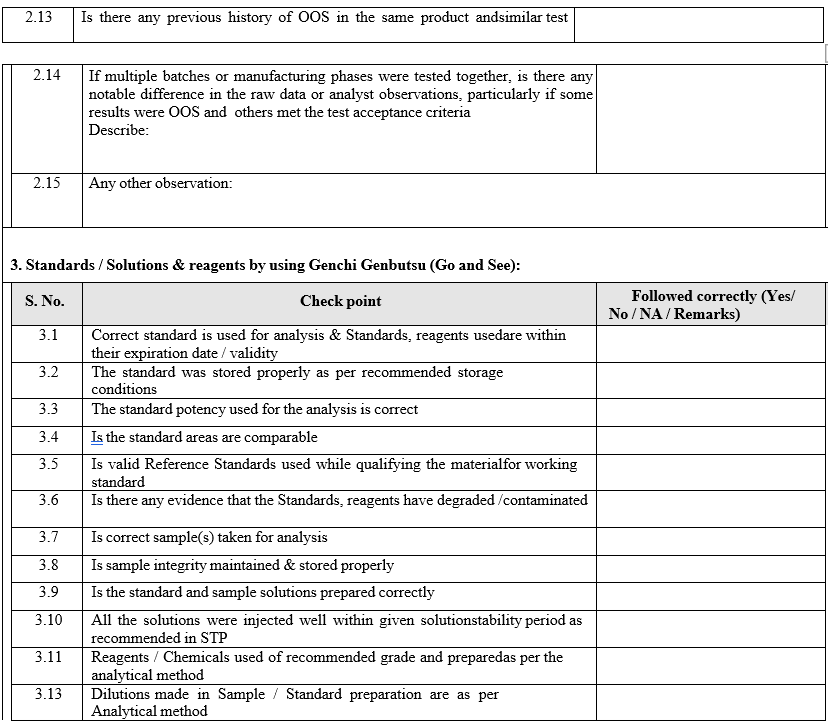
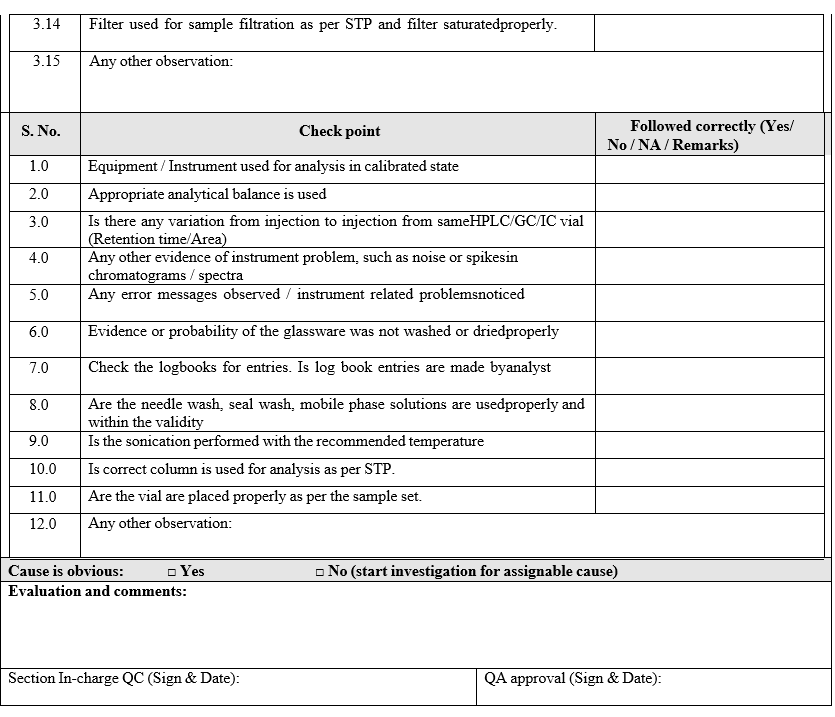
- The Section In charge / designee shall review the raw data, update the check list and forwarded the same to In-charge QA.
- Based on the above review and finding the cause for the OOS may be assigned as a laboratory assignable cause:
Click the link for download word file copy of this document: https://pharmaguidehub.com/product/investigation-of-out-of-specification/
- a. Documentation / transcription error.
- b. Analytical error (related to analyst, method or equipment).
- If the cause is identified as a documentation/transcription error then follow the below procedure:
- Rectify the documentation error/ transcription error.
- Evaluate the document after rectification
- If the batch complies with the specification, then the batch shall be released.
- Even after the rectification of documentation/ transcription error, if the batch does not comply with the specification then continue the investigation.
- If the cause is identified as an analytical error (related to analyst, method or equipment) then follow the below procedure:
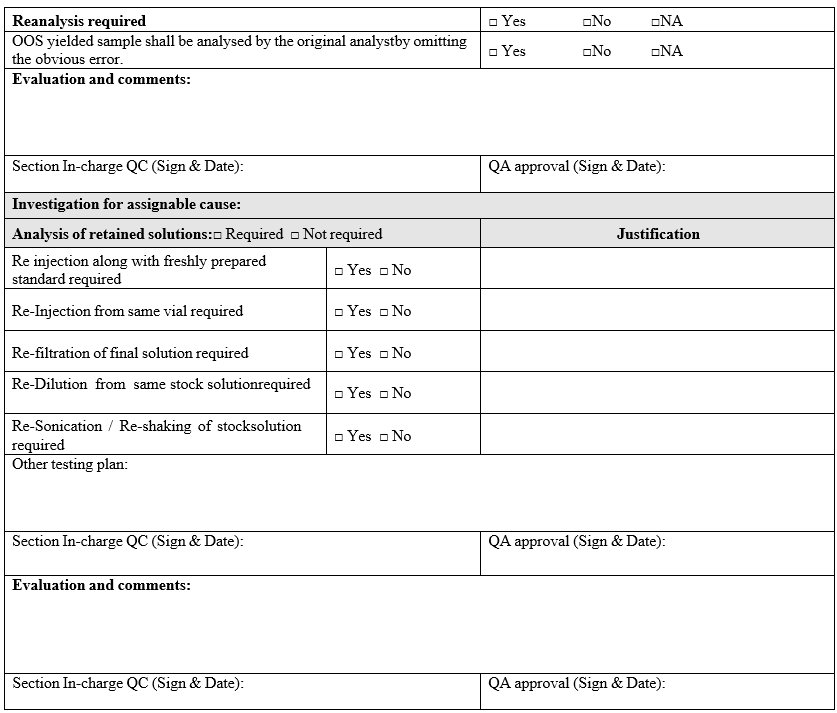
- If the cause for OOS is identified as analytical error and solution stability time has been exceeded for sample aliquot, the OOS yielded sample shall be analysed by the original analyst in single preparation by omitting the assigned error. The reanalysis results shall be considered for reporting.
- If the reason for the OOS is identified as due to the Analyst error like usage of wrong pipette / usage of wrong volumetric flask while making final dilution and solution stability exists for sample aliquot, analysis conditions was not disturbed, then the original analyst shall use existing sample aliquot (e.g., stock solution) and correct dilution shall be prepared and reanalyzed. System suitability must be maintained or re-established.
- In case of any instrument malfunction is identified, information shall be given to instrumentation engineer as per SOP wherever required.
- In all such cases the impact on the samples analysed in the same sequence and previous analysis performed on the same instrument shall be evaluated and documented.
- In the case in which analytical error is related to method instructions, communication shall be given to FARD for revising the method instructions.
- If the batch is complying with the specification, then consider the final results for reporting and forward all the documents along with the OOS investigation report to In-charge QA for evaluation and disposition.
- Even after the rectification of analytical / equipment error, if the batch does not comply with the specification then continue the investigation.
- Note: If the reason for the OOS is identified as due to the Analyst / Documentation / Transcription error then the concerned person shall be counselled/re-trained/demonstrated on the specific procedure or shall be re-qualified on the analytical technique (wherever applicable)
- Analysis of retained solutions:
- Analysis of retained solutions shall be performed to determine probable cause i.e. analysis of retained solutions regarding what might have happened during analysis.
- This analysis of retained solutions may continue from the re-measurement of the original preparations.
- The analysis of retained solutions may not be used to replace original suspect analytical results.
- It may only be used to confirm or discount a probable cause.
- The analysis of retained solutions shall be initiated only after getting prior approval from QA in format-I.
- The justification for performing / not performing the retained solutions testing shall be documented in the format-I.
- Some example of analysis of retained solutions are given below, But not limited to outline the plan for experimental work before starting any activity.
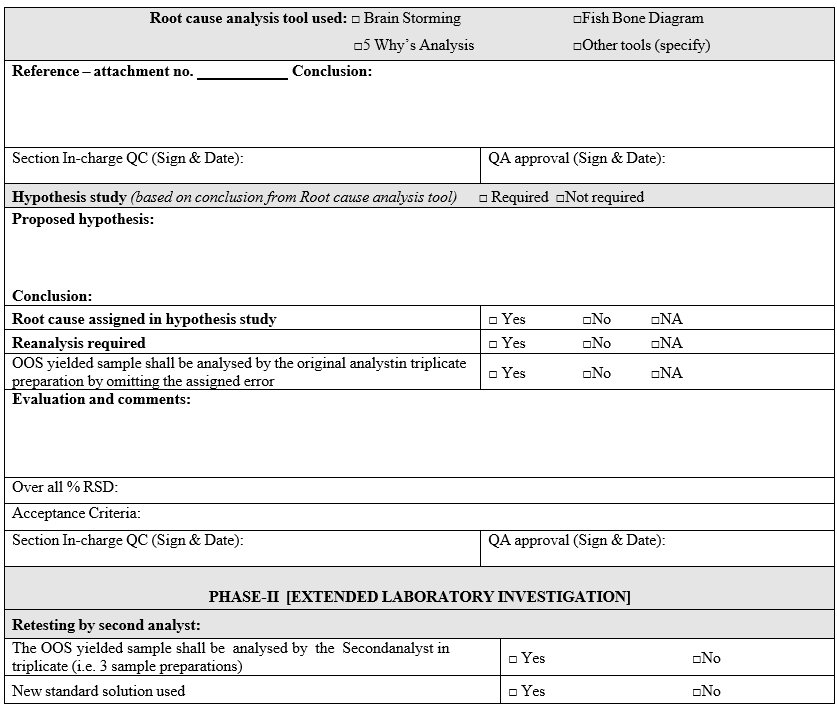
- If the most probable cause is identified during analysis of retained solutions, the investigation for identification of root cause shall be continued by using root cause analysis tools. Root cause analysis tools such as,
- Brain Storming:
- Brain storming is the technique which is a creative event solving method that allows the people to come-up with suggestions / ideas that could solve the event or help to identify the cause of the event.
- It helps to identify potential failure modes and associated hazards, risks, criteria for decisions and/or options for treatment.
- A meeting may be called with cross functional team to brainstorm on the subject event.
- Relevant people shall ask to think and share their views / suggest ideas to overcome the event.
- All views and suggestions shall analyze to identify the cause of the event.
- Fish Bone Diagram (Ishikawa analysis):
- This is a structured method to identify possible causes of an undesirable event or problem.
- It organizes the possible contributory factors into broad categories so that all possible hypotheses can be considered.
- This helps to identify possible root causes, the basic reasons for a specific effect, problem or condition, sort out and relate some of the interactions among the factors affecting a particular process, analyze existing problems so that the corrective actions can be taken.
- 5 Why’s Analysis:
- The strategy involves looking at a problem and asking “Why?”. Very often the answer to the first “WHY?” will prompt another “why?” and the answer to the second “Why?” will prompt another and so on.
- Based on the outcome of root-cause analysis tools applied, a decision shall be made to identify different hypothetical studies to evaluate the most probable root cause identified.
- The analysis of Hypothesis study shall be initiated only after getting prior approval from QA.
- The analysis of Hypothesis study shall be initiated through blank format, shall be issued from by QA.
- Hypothesis study data shall be evaluated to determine the most probable root cause which is scientifically justifiable / meaningful and to identify appropriate corrective and preventive actions.
- If assignable cause identified & confirmed using the root-cause analysis tools, the investigation shall be continued and the OOS yielded sample shall be re-analysed by the original analyst in triplicate preparation by omitting the assigned cause.
- The triplicate results shall meet the acceptance mentioned below:
- Assay – Over all % RSD of three results shall not be more than 2.0 %
- For test Related substances/chromatographic purity- Over all % RSD of three results shall not be more than 10.0 % (applicable only for impurity above 0.1 %)
- For test OVI/Residual solvent- Over all % RSD of three results shall not be more than 10.0 %.
- Content of preservative Over all % RSD of three results shall not be more than 10.0 % If the result of reanalysis in triplicate is not complying with the specification and / or acceptance criteria mentioned above, the investigation shall be continued in Phase-II investigation for non-assignable cause identification.
- In case of Blend uniformity, if assignable cause is identified, the reanalysis shall be carried out with second set of samples by eliminating the assigned error.
- Note: If the reason for the OOS is identified as due to the Analyst, then the concerned person shall be counselled/re-trained/demonstrated on the specific procedure or shall be re-qualified on the analytical technique (wherever applicable).
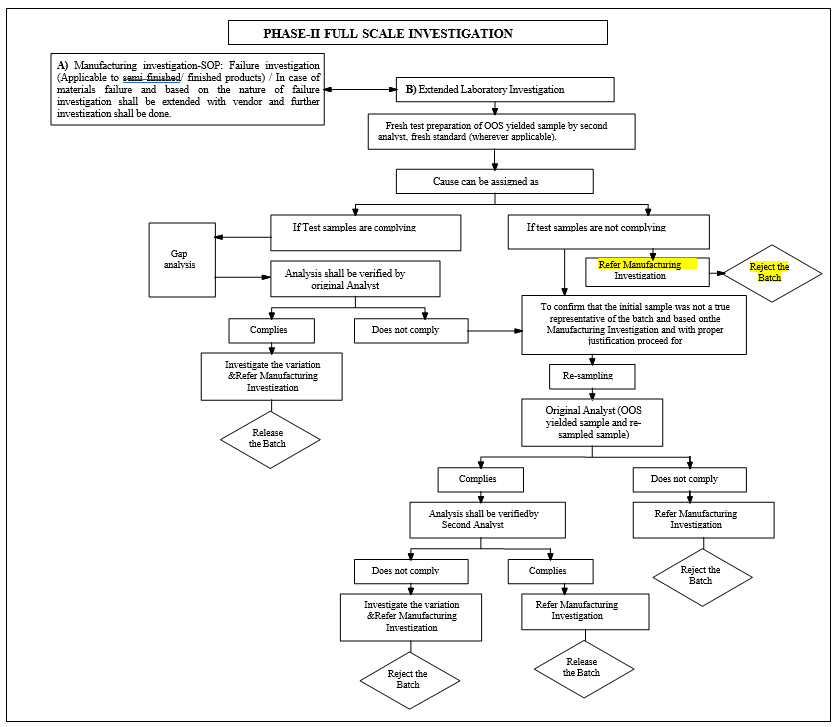
- Phase-II Full Scale Investigation:
- Phase-II investigation includes both Manufacturing investigation and Extended Laboratory investigation which shall be initiated as parallel activity.
- Manufacturing investigation:For Semi finished/finished product and commercialized stability batches the detailed manufacturing investigation shall be performed to find out if there is a manufacturing-related root cause for out of specification. Manufacturing investigation shall be carried out as per procedure in the SOP.
- In case of Drug substances (API, Excipients etc.), following points shall be considered, but not limited to,
- a. The history of the material.
- b. Failure can be shared with the vendor to evaluate and investigate at vendor end.
- c. To substantiate the failure joint analysis also can be planned wherever required.
- In case of exhibit Stability, following points shall be considered, but not limited to,
- a. The trend of the product / test parameter.
- b. Manufacturing/Packing Investigation
- c. Failure can be shared with the FRD / FARD to evaluate and investigate.
- d. After thorough OOS investigation, if OOS is confirmed, based on the recommendations of FRD / FARD, subsequent stations shall be continued. In all such cases OOS need not be initiated.
- Extended laboratory investigation: The extended laboratory investigation includes.
- Retesting of a portion of the original sample.
- Re-sampling.
- Retesting of a portion of the original sample:
- Retesting shall be carried out only when assignable cause is not identified in Phase-I laboratory investigation.
- Reason for retesting shall be documented Retesting shall be carried out after getting prior approval of QA.
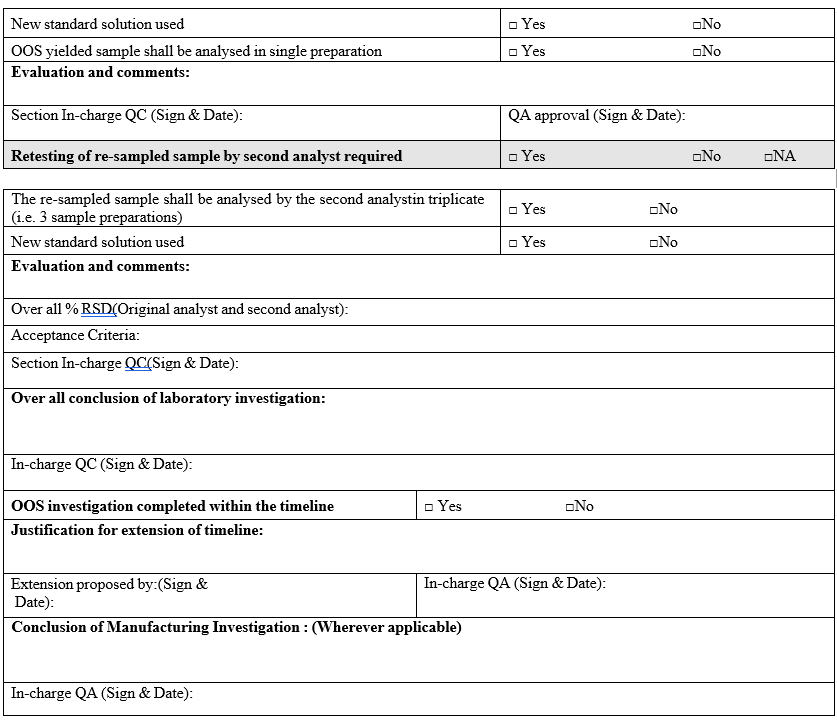
- The OOS yielded sample shall be analyzed by the second analyst (who is equally experienced) in triplicate (i.e. 3 sample preparations) using the fresh standard preparation.
- Retest results complies:
- Gap analysis shall be carried out by the section in-charge to identify the differences in practices followed by second analyst and the original analyst.
- Those differences in practices shall be considered as most probable root causes.
- Analysis shall be repeated by the original analyst in triplicate (i.e. 3 sample preparations) by taking necessary precautions.
- If all individual results by the original analyst are complying with specification, check the reproducibility in results, and verify the compliance with the acceptance criteria as specified:
- Assay –Over all % RSD of six results shall not be more than 3.0 %
- For test Related substances/chromatographic purity-Over all % RSD of six results shall not be more than 15.0 % for “individual impurity” (applicable only for impurity above 0.1 %) and shall not be more than10.0 % for “Total impurities”.
- For test OVI/Residual solvent- Over all % RSD of six results shall not be more than 15.0 %.
- Content of preservative – Over all % RSD of six results shall not be more than 15.0 %
- Average the all the test results and substitute average value for the initial test results of the original analyst. (Before taking the decision for Rejection or Release of batch, Manufacturing Investigation shall be taken into consideration.)
Click the link for download word file copy of this document: https://pharmaguidehub.com/product/investigation-of-out-of-specification/
- Retest results not complies:
- If any individual result of OOS yielded sample is not complying with the specification, then the investigation shall be extended to confirm the initial sample integrity by re-sampling.
- The investigation shall be extended to confirm the initial sample integrity by re-sampling. Continue the investigation as per re-sampling procedure mentioned above.
- Based on manufacturing investigation, if the possibility of sample integrity is not obvious the batch shall be rejected after evaluation.
- In-charge Quality control shall take the decision for further investigation in association with In-charge Quality assurance.
- In case of Blend uniformity, if assignable cause is not identified, the retesting shall be carried out with second set of samples.
- Re sampling:
- If any individual result is not complying; investigation shall be extended to confirm the initial sample integrity by re-sampling.
- To confirm the sample integrity of the initial OOS yielded sample and to verify that the initial OOS yielded sample was not the true representative of the batch, In-charge-QC shall recommend for the re-sampling with reason and proper justification.
- Re-sampling shall be approved by In-charge –QA. (Note: Re-sampling shall be performed only when potential sampling error has been identified during Manufacturing Investigation and/or to verify the initial sample was a true representative of batch or not. Re-sampling shall be performed by the qualified and validated method.)
- The re-sampled sample shall be tested by original analyst in triplicate with using the fresh standard preparation along with OOS yielded sample.
- If any individual result of re-sampled sample by original analyst is not complying with the specification, then the batch shall be rejected. (Before taking the decision for Rejection or Release of batch, Manufacturing Investigation shall be taken into consideration.)
- If all individual result of re-sampled sample by original analyst are complying with the specification and results for OOS yielded sample represents the same OOS yielded results, then the analysis shall be verified by the second analyst in triplicate.
- If any individual result of re-sampled sample by second analyst is not complying with the specification, then the batch shall be rejected. (Before taking the decision for Rejection or Release of batch, Manufacturing Investigation shall be taken into consideration.)
- If all individual results of re-sampled sample by second analyst are complying with the specification, then the batch shall be released by considering final average results of original and second analyst.
- Note: Detailed investigation shall be carried out to find out the variation in results. (Before taking the decision for Rejection or Release of batch, Manufacturing Investigation shall be taken into consideration.)
- If the investigation is inconclusive and a cause for the OOS result cannot be assigned, the investigation shall be closed and all the investigation details shall be sent to In-charge – QA for the disposition of OOS.
- Head QA or QC shall take the decision for additional investigation and/or to hand over the investigation to other concerned departments such as FRD / FARD for further investigation.
- Impact Assessment:
- In case of any instrument malfunction is identified, the impact on the previous analyses performed on the same instrument shall be evaluated and documented.
- In case OOS observed in batches in a sample set, the other batches analysed together shall be evaluated for the impact before release.
- The impact of OOS results on other batches, ongoing stability studies, validated processes and testing procedures shall be determined by Head QC and Head QA and be documented in the conclusion, along with appropriate corrective and preventive actions.
- The impact on prior analyses shall be evaluated if the cause is assigned as the method.
- If a similar kind of OOS was observed earlier for other batches of same product/strength; for similar test parameter, the reference of previous OOS investigation shall be considered during investigation.
- A comprehensive served in the same product / strength for similar test parameter for 3 occurrences, a comprehensive investigation summary shall be performed along with necessary CAPA.
- If result was expected on the basis of trend available for the batch / product, action shall be taken as mentioned below:
- Head QC and Head QA shall perform a critical evaluation of data to conclude that result was really expected.
- If as an outcome of evaluation it is confirmed that result was expected, no further laboratory investigation is required.
- Results shall be shared with FARD and FRD for further investigation.
- In case Specification is revised as a part of investigation, study shall be continued with new specification.
- If it is revealed that OOS results obtained due to problem with analytical method and method is revised as a part of investigation, Analysis shall be performed with new method.
- If specification / method are not revised, study shall be stopped at accelerated condition and study at intermediate condition shall be initiated.
- Analysis of accelerated condition samples (40°C/75% RH) to be continued up to 6 month irrespective of failure / OOS observed during stability for exhibit batches of US Market only.
- In case of Intermediate and long term condition, decision for study to continue / discontinue shall be taken by FARD, FRD, CQA & QA departments.
- All original data shall be retained and proper scientific justification shall be documented.
- Out of specification investigation shall be closed within 25 working days. If the investigation has to extend beyond 25 days, the reason for extension of the investigation shall be reported.
- If any additional investigation/additional testing, other than the procedure mentioned above is required, the same shall be recorded, justified and approved.
- If the OOS is confirmed, batch shall be rejected and out of specification log shall be subsequently updated with the Investigation disposition status and closing date.
- Once the batch has been rejected there is no limit to further testing to determine the cause of failure, so that corrective action shall be taken.
- The decision to reject cannot be reversed as a result of further testing.
- QA-Executive shall initiate the MNC as per SOP.
- As per the quality agreement (wherever applicable) the out of specification results shall be intimated to concern Qualified Person of the customer.
- Market complaints or returned caused due to an out of specification result shall be investigated as per SOP.
- In case of the OOS investigations for stability following points shall be taken into consideration and extend the laboratory investigation:
- Trend of the product
- Nature of the failure
- History of the failure
- Expected failure
- Comparison between other strengths and packs
- Comparison between similar manufacturing formula
- Pack integrity
- Condition specific
- In order to complete the laboratory investigation, the section in-charge/designee and analyst may take into consideration the following facts:
- Expected percentage of error for the particular test, if available. Results obtained for the particular lot in other tests or release parameters (evaluate overall behavior of lot) sometimes the value is reproduced or substantiated by other analytical tests.
- Evaluate history of particular product and validation results, if available. Process capability and trend analysis may also help to complete investigation and clarify results.
- Enough documentation should be provided in order to evidence that an OOS value belongs or does not belong to the set of data obtained for the particular lot.
- Averaging in OOS results:
- Averaging results from a standard or test aliquot is acceptable (i.e. averaging replicate results).
- Averaging a set of results, where some are OOS is not acceptable.
- Hiding an OOS result in any average is not acceptable.
- In case the test is related to investigate the variability of product then averaging is not acceptable in such cases and RSD values shall be reported to show statistical significance.
- Calculation of an outlier:
- If overall % RSD is within the defined acceptance criteria and the value of the test is different from the others in a series such value may be an outlier.
- Outlier is not applicable to content of uniformity, dissolution and/or release-rate determination.
- Outlier is not applicable to chemical test.
- Outlier shall be considered only during the additional testing performed in an OOS investigation, the laboratory will obtain multiple results.
- Handling of OOS results in Physical tests:
- Incase of any discrepancy observed w.r.t. physical appearance like description, appearance, colour, non-conformance with approved reference shade card/art-work etc., the material/product shall be verified on another fresh pack from investigation sample/control sample / reference sample if available. Further retesting shall not done, however analysis of other parameter shall be carried out, if required.
- The material/product shall be rejected / approved by QA and further investigation shall be done with respective SOP.
- QA-Executive shall initiate the MNC as per SOP.
- Handling of OOS results in Microbiology tests:
- Incase of microbial tests like Microbial Limit tests, Bioburden etc., OOS notification and investigation shall be carried out as per SOP
Click the link for download word file copy of this document: https://pharmaguidehub.com/product/investigation-of-out-of-specification/
- The following criteria shall be considered prior to replacing the initial OOS results:
- Obvious error identified during laboratory investigation.
- Most probable root cause is identified and confirmed during analysis of retained solutions and hypothetical studies.
- Review of data related to other stages and tests.
- Review of manufacturing investigation where investigation is extended to Phase-II.
- Review of product history.
- Only when a clear evidence of laboratory error exists, initial testing results should be disregarded.
- Trending of Out of Specifications:
- QA shall prepare trends of OOS on quarterly and annually to determine the effectiveness of the system and identify area of improvement. Trend of OOS should include at minimum;
- Number of OOS.
- Assignable cause.
- Root Cause.
- Number of OOS per Product/Material.
- Number of confirmed OOS.
- Overdue OOS investigations.
- Trend analysis should identify any methods, materials, and equipment or personnel errors consistently occurring. Appropriate, corrective action shall be initiated.
- Trend analysis should identify particularly those OOSs which were attributed to Laboratory errors and perform in-depth investigations (such as review of analytical methods etc.) to identify the root causes for Laboratory errors and implement corrective actions.
- Trend analysis shall be documented and shall include recommendations for corrective actions and distributed to appropriate department heads.
- Based on final evaluation and comment, corrective and preventive action shall be proposed to prevent re-occurrence of OOS.Continuous improvement actions that do not require investigation may be handled as per the CAPA procedure.
- Trend reports shall be reviewed by Executive/Designee QA and Head QC.
- Trend reports shall be approved by head QA.
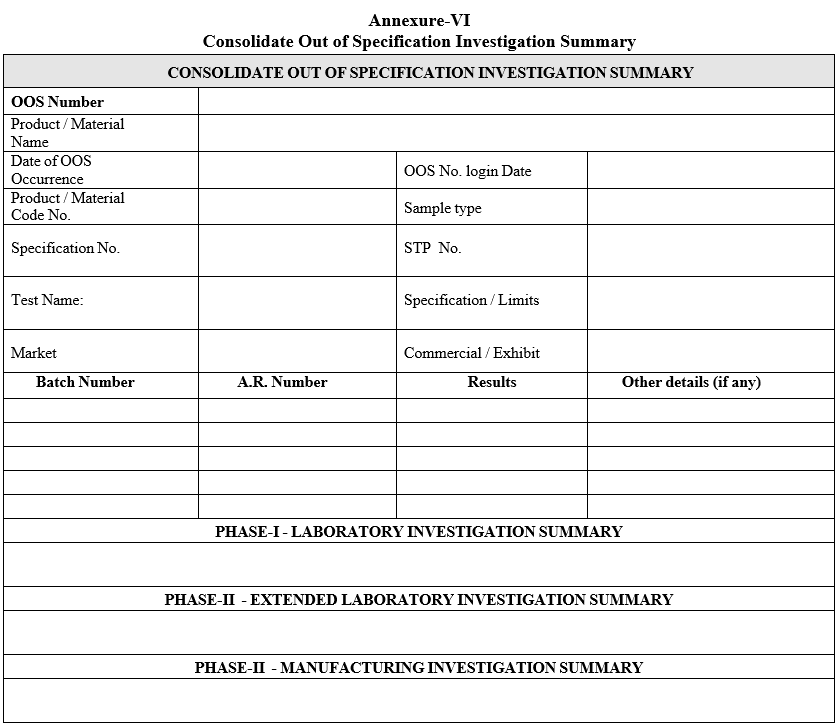
- A Consolidate Out of Specification Investigation Summary (including laboratory and manufacturing investigation) shall be prepared as per format-V in case of both the laboratory and manufacturing investigations are involved.
Definations:
- Out-of-specification test result: Test result that does not comply with the pre-determined acceptance criteria.
- Laboratory Assignable cause: An error associated with the performance of a test procedure or due to analyst, laboratory equipment malfunction or failure.
- Assignable cause: A scientifically justified explanation of the reason for an out of specification or questionable test results uncovered and documented during the laboratory investigation.
- Reanalysis: In case of assignable laboratory error, analysis to be carried out on the same test solution or freshly prepared sample composite from the original sample.
- Retest: In case of non-assignable laboratory error, analysis to be carried out on the freshly prepared sample composite from the original sample or re-sample.
- Resample: A second or additional composite sample collected from a lot or batch after drug substance or drug product by following a standard sampling procedure.
- Hypothesis/Investigative Testing: A testing to be performed to confirm or discount a possible root cause i.e. what might have happened that can be tested.
- Obvious error: The error is clearly identifiable e.g.: wrong glassware usage, wrong method used, wrong samples usage, spilling of the sample solution, incomplete transfer of a sample etc…
- Phase I investigation: Initial investigation conducted by the analyst and section In-charge / designee to identify obvious / assignable laboratory error.
- Phase II investigation: The investigation conducted when the phase-I investigation did not reveal an assignable laboratory error.
- Second analyst: The analyst should be at least as experienced and qualified in the method as the original analyst.
- REFERENCES:
FDA guidance for industry: Investigating out of specification test results for pharmaceutical production
- ANNEXURES:
| ANNEXURE NO | TITLE OF ANNEXURE |
| Annexure-I | Out of specification Investigation summary report |
| Annexure-II | Out of specification Investigation Logbook Quality control |
| Annexure-III | Out of specification Investigation logbook Stability |
| Annexure-IV | Analyst Training |
| Annexure-V | Out of Specification Laboratory Investigation Report |
| Annexure-VI | Consolidate Out of Specification Investigation Summary |
- ABBREVIATIONS:
| No. | : | Number |
| SOP | : | Standard Operating Procedure |
| OOS | : | Out Of Specification |
| FAR | : | Field Alert Report |
| RSD | : | Relative Standard Deviation |
| FRD | : | Formulation Research &Development |
| MNC | : | Material non conformance |
| API | : | Active pharmaceutical ingredient |
| QC | : | Quality Control |
Out of specification Investigation flow chart
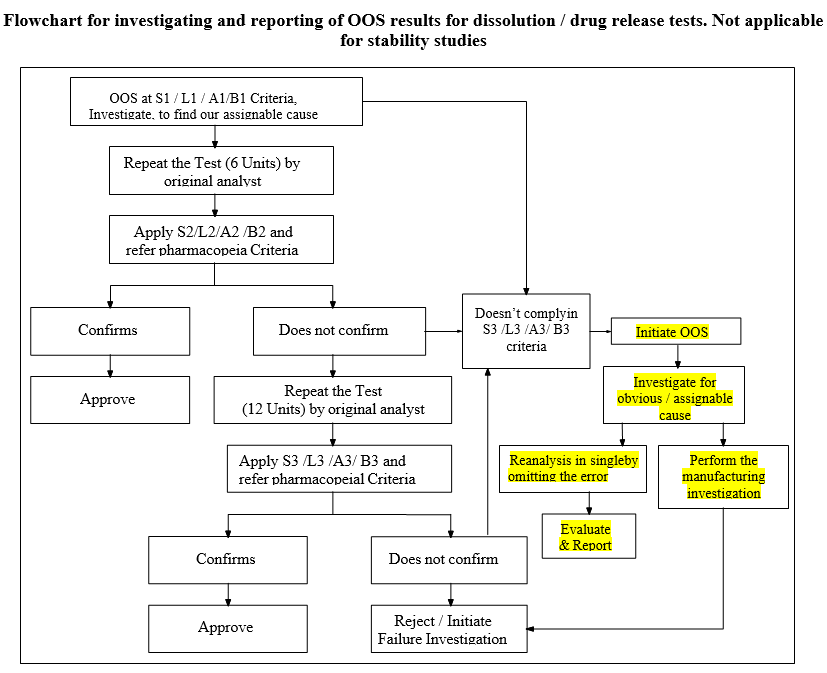
Flowchart for investigating and reporting of OOS results for dissolution / drug release tests. Not applicable for stability studies
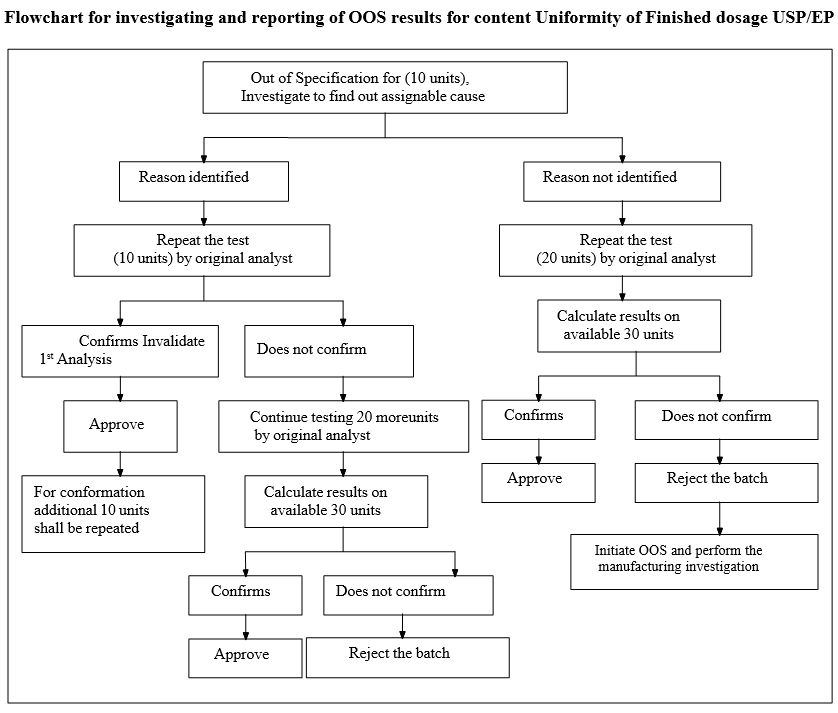
Flowchart for investigating and reporting of OOS results for content Uniformity of Finished dosage USP/EP
Annexure-I
Out of specification Investigation summary report


Click the link for download word file copy of this document: https://pharmaguidehub.com/product/investigation-of-out-of-specification/
Annexure-II
OUT OF SPECIFICATION INVESTIGATION LOGBOOK QUALITY CONTROL
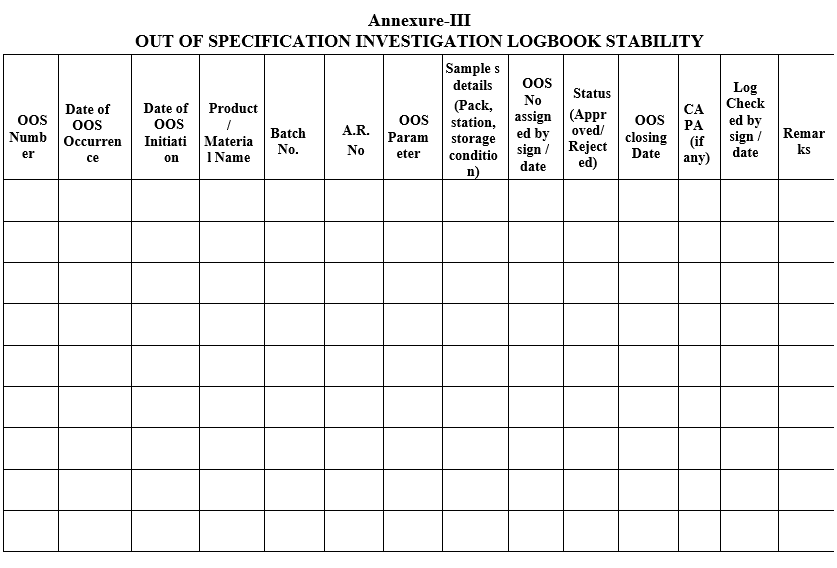
Annexure-III
OUT OF SPECIFICATION INVESTIGATION LOGBOOK STABILITY
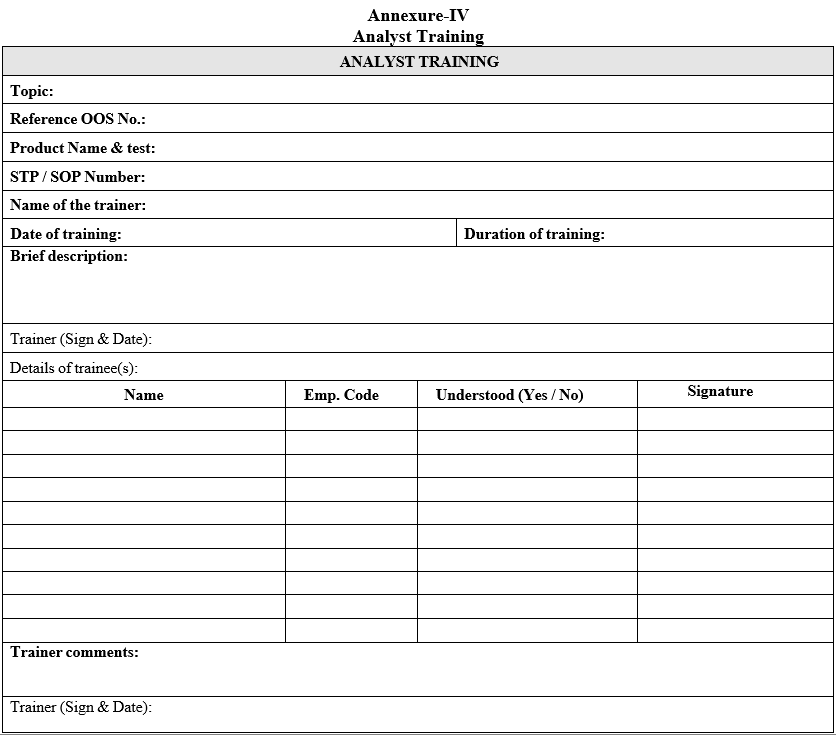
Annexure-IV
Analyst Training
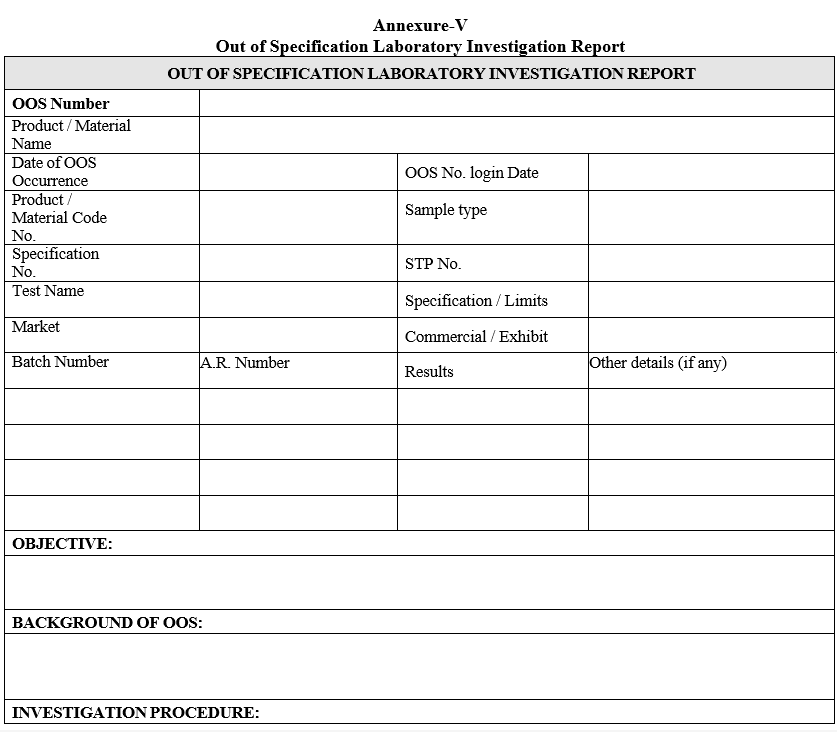
Annexure-V
Out of Specification Laboratory Investigation Report

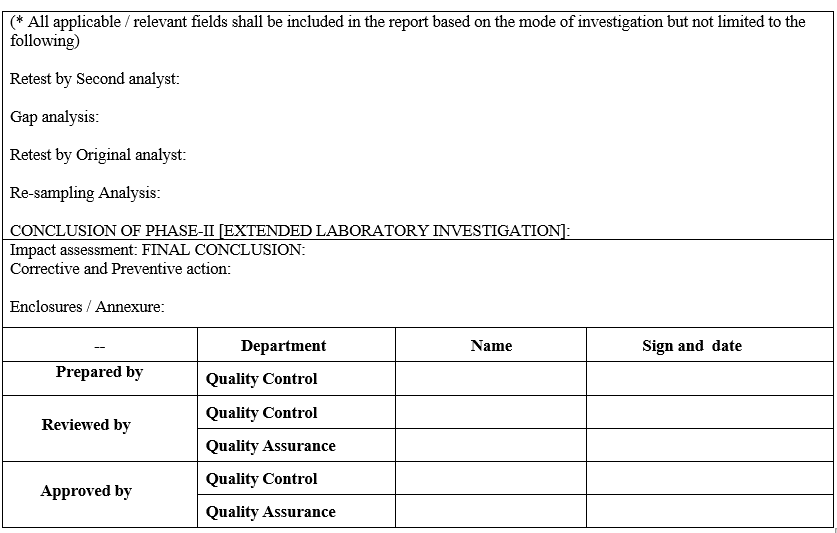
Annexure-VI
Click the link for download word file copy of this document: https://pharmaguidehub.com/product/investigation-of-out-of-specification/