

Objective
To provide a procedure for investigation of out of specification (OOS) result found during the testing of raw materials, in- process samples, finished products, complaint samples and stability samples.
Scope
This SOP is applicable for investigation of OOS results of all tests performed on raw materials, in- process samples, finished products, complaint samples and stability samples within their expiration date for chemical analysis only, at {Company Name} {Location}.
This SOP is applicable for in case of OOS results in test “Dissolution” (After confirmation at stage criteria as per respective pharmacopeia (e.g. after S1, S2 and S3 level not meet the criteria etc.) and “Uniformity of dosage units” (After confirmation at L1, and L2 level).
This SOP is not applicable for :
Microbiological testing.
Testing of environmental monitoring samples.
Test performed during product development, method development and Analytical method transfer.
Analytical data generated by analyst at the time of training and analyst qualification.
Packing Material i.e. carton, foil shipper etc. same shall be covered through incident/Deviation.
Test of physical parameter e.g. Description, Average Weight, Thickness, Hardness, and Friability etc. same shall be covered through incident/Deviation.
Stress study testing conducted at elevated/adverse exposure conditions intentionally done to check an impact on the characteristics of material/product e.g. force degradation study.
Responsibility
Analyst
Responsible for identifying the OOS results and provide the relevant data, information/ documentation to support the basis of OOS and immediately reporting to section in-charge. Report suspect results to a Supervisor.
Responsible for retain all the preparations, portion of test sample, reagents glassware used in analysis when unexpected or OOS results are obtained.
Responsible to provide the support to section in-charge during preliminary investigation. Determine the course of action and propose experimental hypothesis, with the assistance of a Section In-charge.
Section In-charge
Responsible to inform QA department for maintaining the OOS log and issuance of the format.
Responsible for immediate assessment of OOS results that include the re- examination of actual solution, test units and glassware used in original measurement and preparation, which might provide more credibility for laboratory hypothesis. Discuss the test method with analyst; interview the analyst for performance of the correct procedure.
Responsible for evaluation of the performance of the instruments and test method to ensure performance according to the standard expected based on the method validation. Evaluate the performance of the test method to ensure that is performing according to the standard expected based on method validation data and historical data
Responsible for conducting preliminary laboratory investigation with analyst as per checklist.
Responsible to conduct the investigation within 3 business day.
Identify causes, initiate corrective/preventive actions.
QC Head
Responsible for review and confirming the accuracy of OOS result.
Responsible for evaluation/investigation of OOS with respect to laboratory aspects of reported OOS result.
Responsible for compliance of this SOP.
Responsible to give the necessary inputs for Phase-II investigation, if root cause of OOS result does not identified during phase-I investigation.
Responsible for CAPA (if laboratory error identified) to avoid the recurrence of similar OOS.
QA Head/Designee
Responsible for review of laboratory investigation report, production investigation report and provide his recommendations/conclusion for same.
Responsible for review of laboratory investigation report, production investigation report and provide his recommendations/conclusion for same.
Responsible for suggesting necessary CAPA and informing to other department as applicable.
Responsible for developing strategy for inter departmental investigation (i.e. full scale investigation) that may imitated as a result of an inconclusive preliminary laboratory investigation.
Responsible to give the authorization for additional laboratory testing (re- sampling/ retesting plan).
Responsible to initiate the appropriate controls e.g. quarantine, operation as may be warranted until an investigation shall be completed.
Responsible for approval of OOS investigation report and reject/release/Disposition the batch based on the OOS investigation report.
Production Head/Engineering Head/ Warehouse Head:
Responsible for performing full scale investigation including shop floor investigation with QA department.
Responsible for CAPA if error identified during process related investigation, to avoid the recurrence of similar OOS.
Accountability
Head QA and Head QC shall be accountable for the compliance of this SOP.
Procedure
General conditions
In case of any results goes out of specification limit during analysis, the analyst shall immediately inform to section in-charge / Head QC or designee. Before discarding test preparations or standard preparation analyst shall check the data for compliance with specification.
When OOS results are obtained and no obvious explanation exists, test preparation shall be retained by the analyst (Keep all solution on bench with marking “Do not Discard”) and inform to the Section Incharge (QC) or designee.
The analyst shall document the errors, if happened, during the analysis such as spilling of samples, solutions, incomplete transfer of composite samples etc. In such cases he/She shall not continue an analysis of the prepared sample solutions.
After an OOS is reported the Head-QC or designee shall immediately assess the accuracy of the results/data.
Section Incharge (QC) or designee shall initiate an OOS by requesting QA for issuing the required documents.
OOS result can be due to
Assignable cause (Laboratory error or manufacturing process error or non-process related error or operator error).
Non Assignable cause
The investigation shall be thorough, timely, unbiased and documented.
An OOS investigation shall be completed within 30 working days.
In case the investigation exceeds more than 30 days then interim report shall be generated by QC manager/designee and extension shall be taken for investigation after authorization by Quality Assurance Head/Designee.
Wherever applicable Quality Assurance head shall notify the OOS results to head of concerned RA, Manufacturing Head/customer as defined in respective agreement.
The purpose of investigation is to determine the cause of the OOS, even if a batch is rejected based on an OOS result. OOS investigation may be conducted (if required) if the result is associated with other batches of the same product or material or other products or other materials. Batch rejection does not negate the need to perform investigation.
For an OOS obtained during contract testing, investigation carried out by contract testing laboratories, this laboratory shall initiate a full-scale investigation. The responsibility of a contract testing laboratory in meeting cGMP requirement is equivalent to that of a manufacturing firm. Manufacturing firm shall ensure the availability of adequate OOS procedure in place at contract laboratory. Laboratory shall report its finding and shall provide all supporting documents to the sender unit laboratory.
The analyst shall be aware of potential problems that could occur during the testing process.
The analyst shall ensure that only those instruments/equipments are used for analysis which shall meet established specifications and all instruments/equipments are properly calibrated/ validated.
The investigation shall include an initial assessment of the accuracy of the laboratory’s data. Whenever possible, this shall be done before test solutions are discarded.
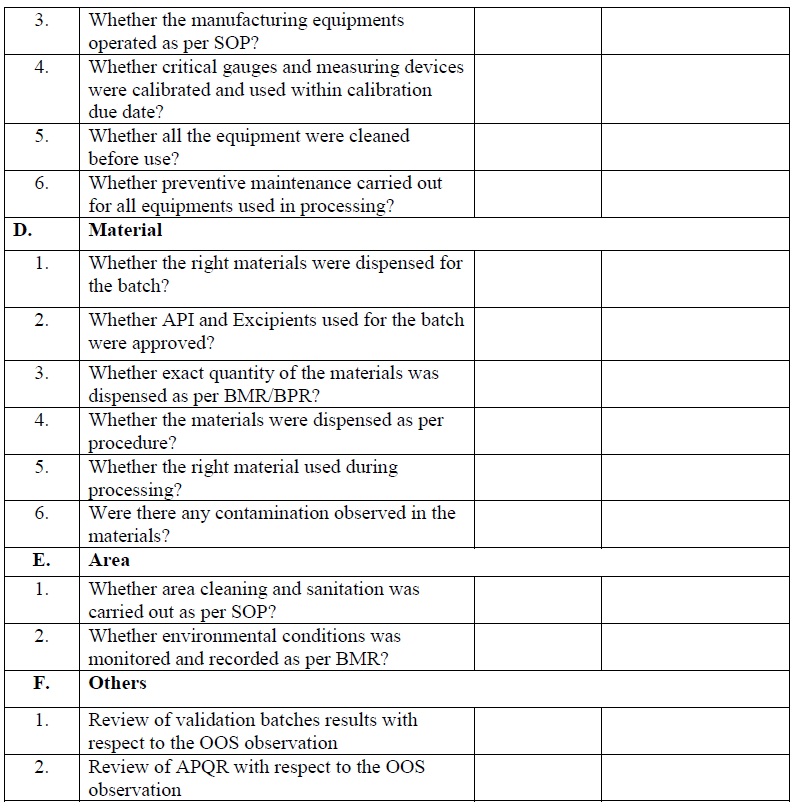
Following procedure shall be adopted for different types of materials.
Raw material: Phase-I investigation shall be carried out and wherever applicable, if assignable cause is not identified then QA Head/ Designee shall discuss the information with purchase/R&D/RA/CQA and vendor regarding the OOS result of material and shall proceed on the basis of outcome.
In-process samples: Phase-I and Phase-II investigations shall be performed.
Finished product: Phase-I and Phase-II investigations shall be performed.
Stability sample: Phase-I and phase-II investigation shall be performed. If OOS is observed in subsequent time point of the same batch of same product then in such case Phase-II investigation report of initial reported OOS shall be referred.
On obtaining the OOS result analyst who performed the initial test shall immediately inform the OOS result to section in-charge.
Section in-charge shall inform to QA department for maintaining the OOS log and investigate the OOS result as per check list (Phase-I investigation).
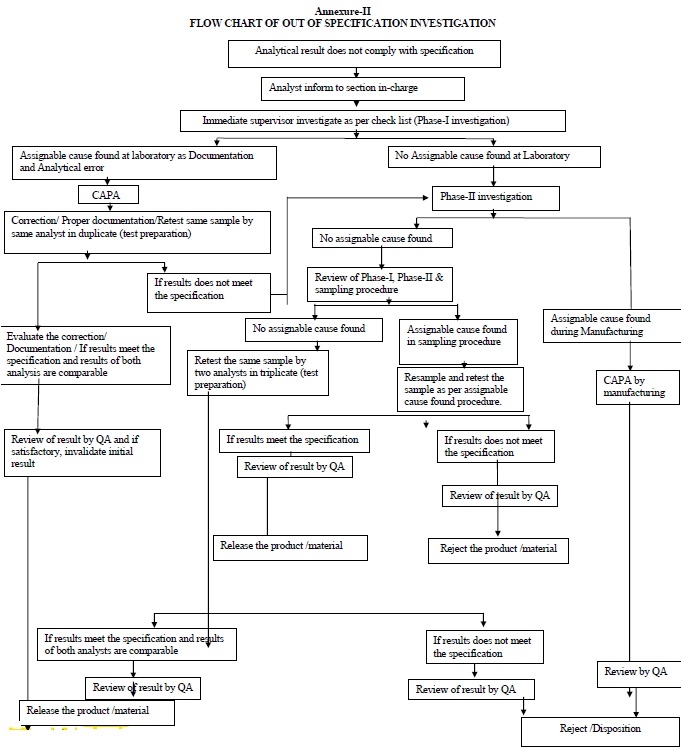
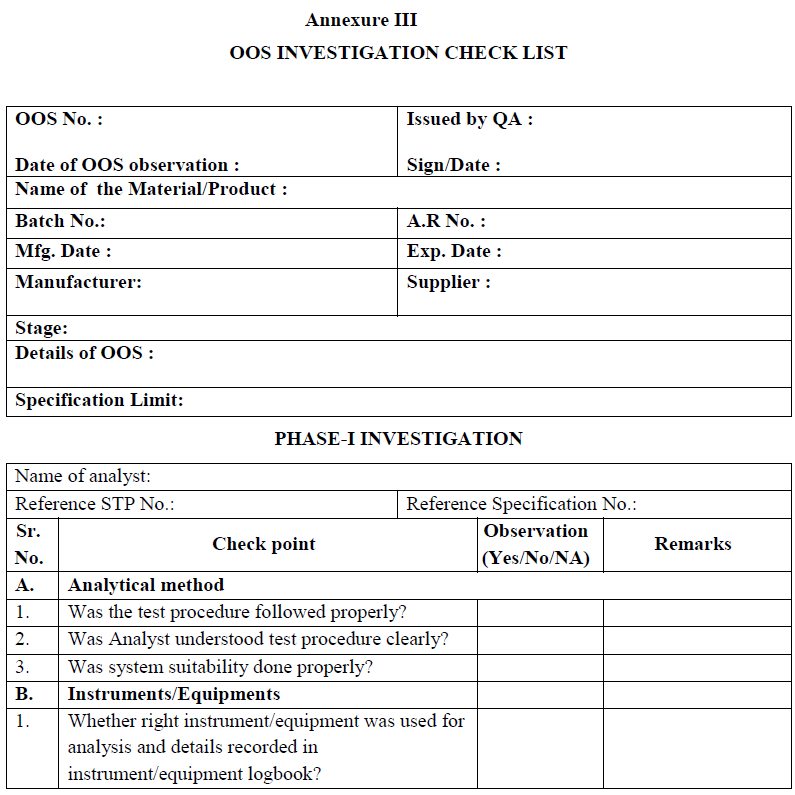
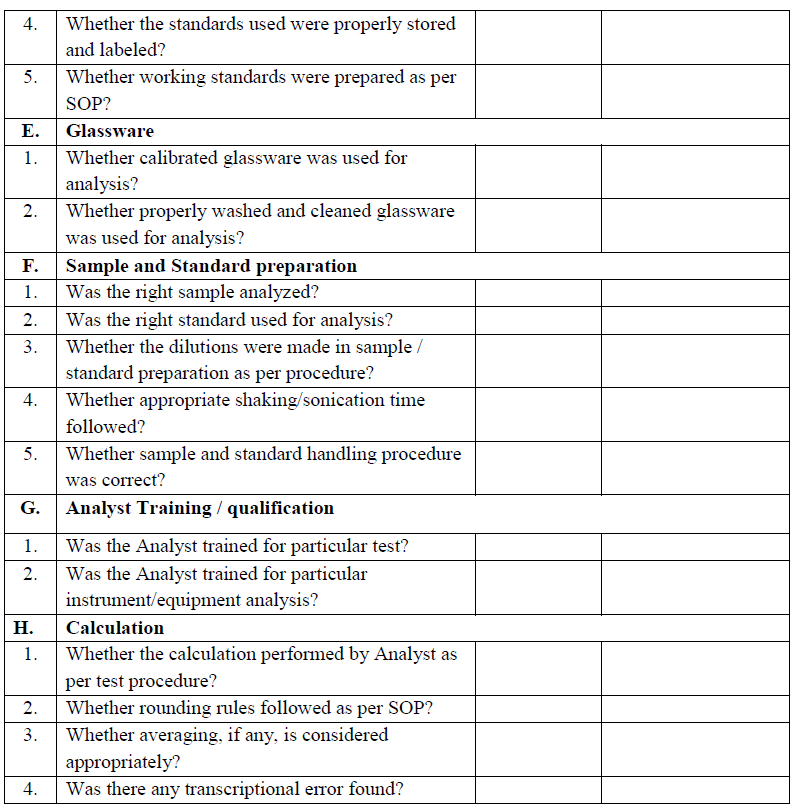
Phase-I Investigation (Preliminary laboratory investigation):
This investigation is to determine if there is an error in the laboratory during testing and has no impact on the specification of the product manufactured. The first phase of a laboratory investigation (Phase-I) is to determine if the proper methodology, instrumentation and calculations were used. In this phase obvious laboratory errors are investigated. The Phase-I investigation shall include an interview with the analyst who performed the testing that lead to the OOS result, and completion of the appropriate
investigation checklist. This phase may include re-measurements of the original working solutions, including re-injections and re-dilutions of the original solutions. If there is clear evidence that a laboratory error occurred, then the original result shall be invalidated at the conclusion of phase-I. For these instances a Phase-II investigation shall not be performed.
Whenever OOS result is observed, analyst shall preserve all standard and test solution as well as reagents which is used for analysis, whenever possible. The same shall be discarded after conclusion of Phase-I investigation.
Section in-charge shall discuss with the analyst, regarding the execution of entire analytical procedure, calculations used, analytical knowledge and competency, and shall proceed investigation as per check list.
Section in-charge shall examine the raw data obtained in the analysis, including chromatogram and spectra to identify anomalous or suspect information. Examination may include a review of the following Points :
- Method validation data for solution stability and recovery levels.
- Analytical method for its suitability.
- Trend of analytical data.
- Result of other batches, which were run concurrently.
- Check analyst qualification record.
- Check OOS recorded by analyst in Past history.
- Check whether the same nature or OOS has been observed or recorded in previous history for the same product.
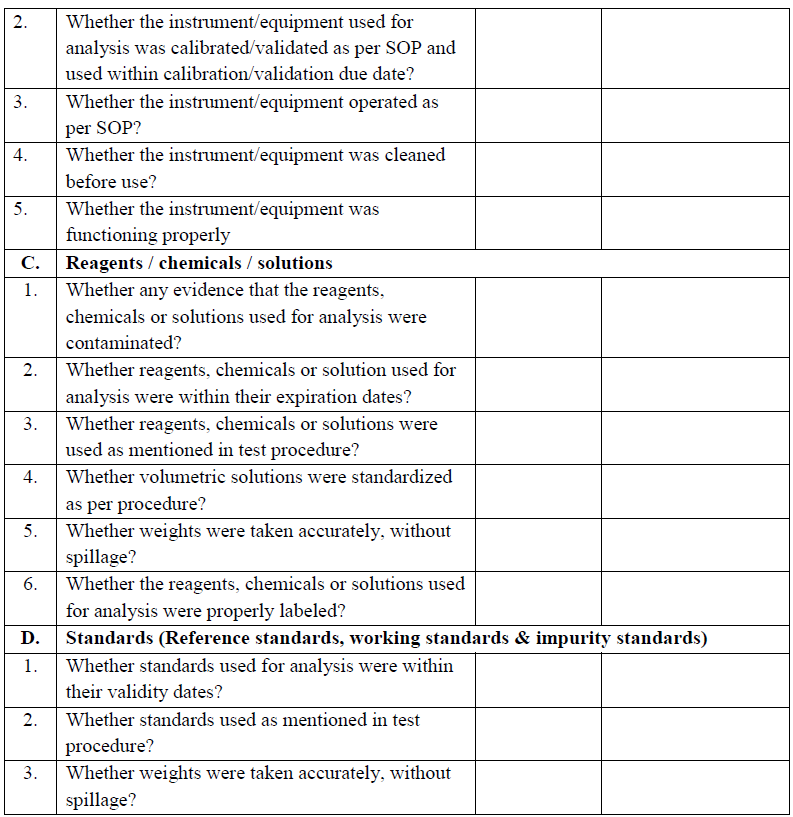
- Verify and confirm the performance of the instrument/Equipment Calibration, validation, maintenance, cleanliness, system suitability.
- Inspection of work area to determine if any environmental or facility conditions would have adversely impacted the testing.
- Identification and evaluation of unusual events or circumstances associated with testing.
Laboratory investigation shall include but are not limited to the following :
- Analyst error.
- Calculation error.
- Weighing error.
- Dilution error.
- Instrument performance error.
- Non compliance to critical steps during the analysis.
- Reference standard/working standard used in the analysis expired or its assay value is incorrectly determined.
- Expired volumetric solutions or reagents usage.
- Improper sample handling during analysis.
During Phase-I investigation one of following outcome may be recorded.
- Error resolved with retest: This appears where an error is clear and can be corrected, then proceed as per assignable cause.
- If no laboratory error is observed and no suspected cause is found then proceed directly to Phase-II investigation.
During the laboratory investigation (Phase-I investigation), following steps can be proceed to identify the root cause of OOS result.
- If re-analysis is necessary to deemed as outcome of preliminary laboratory investigation then it is to be performed on original sample preparation solution which was tested and yielded the OOS result
- Sample solution can be further diluted from retained stock solution to confirm the dilution error and final sample solution can be rejected. Re-injection of retained solution preparation to confirm instrument malfunction. Such hypotheses are difficult to prove. However, re-injections can provide strong evidence that the problem shall be attributed to the instrument, rather than the sample or its preparation.
- Wherever the sample is planned to be re-injected, the stability of the sample solution for the period (for the duration of the period from the initial observation to the planned re-injection time) need to be supported by the data generated during the validation of method.
- Re-filtration of sample solution can be investigate if OOS result is due to filtration error such as improper filtration, in appropriate filter paper used, insufficiently filter paper or contaminated vials/filter. Same dilution of sample solution shall be re-filtered (taking all necessary precaution) and analyzed.
- Re-sonication or re-shaking can be done if it is appear that OOS result is due to incomplete solubilization of analyte in the test preparation due to inadequate sonication or shaking of test solution.
- For release rate testing of certain specialized dosage form drugs that are not destroyed during testing, where possible, examination of the original dosage unit tested might determine whether it was damaged during laboratory handling in a way that affected its performance. Such damaged shall provide evidence to invalidate the OOS test result and retest shall be indicated.
- Further extraction of dosage unit to confirm the complete extraction.
Whenever a laboratory error is identified and OOS result was caused by the testing laboratory, then the source of that error shall be determined and appropriate corrective action and preventive action shall be taken to prevent the re-occurrence of OOS results.
If the OOS result occurred due to an analyst error, training shall be given to the concerned analyst and training report to be attached with OOS report.
Section in-charge and QC Head will evaluate previous data for similar type errors and the potential impact on previously reported results. For example if analyst has diluted a solution 25ml instead of 20ml, which result in lower value, previous analysis by same analyst shall be reviewed for similar type error. Impact evaluation, corrective and preventive action is to be documented in laboratory investigation report.
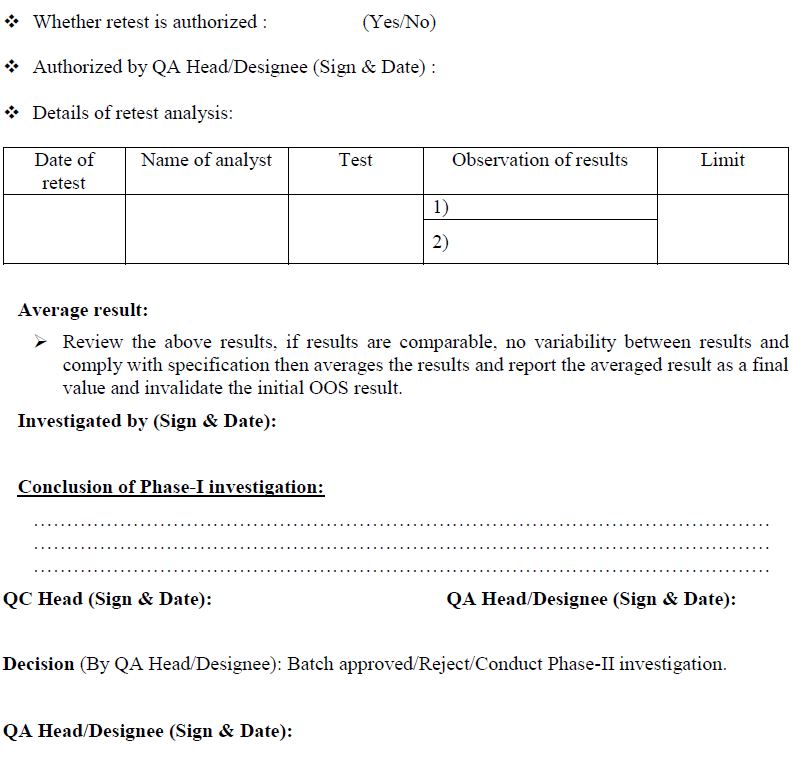
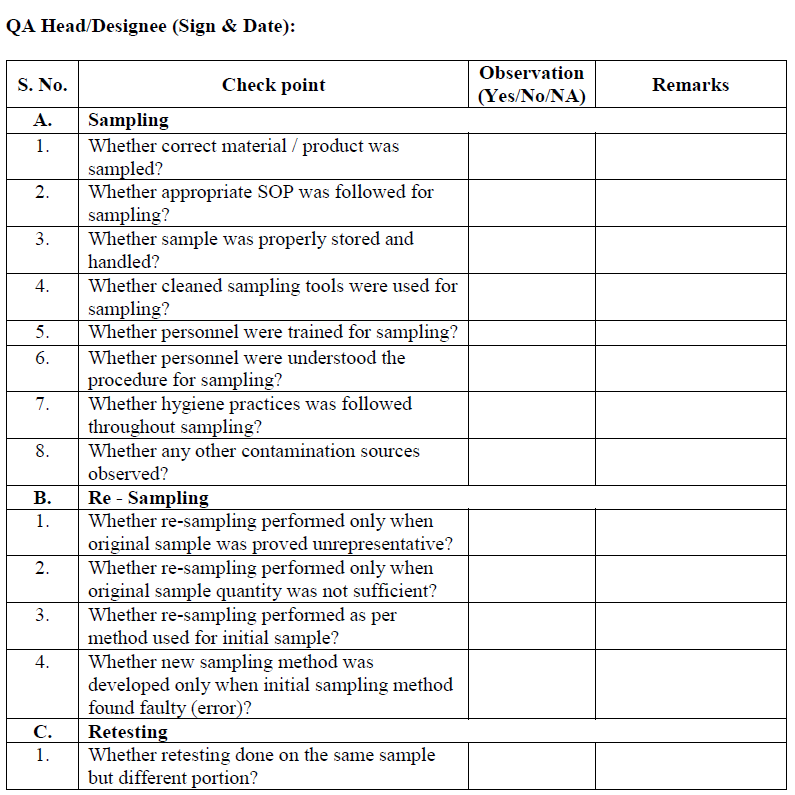
Retesting for Assignable cause identified:
If assignable cause found & laboratory deviation or error is identified after Phase-I investigation then after approval of QA Head/ Designee, perform the retesting of sample by original homogenous sample that was initially collected from the lot or batch that yielded the OOS result. It may be additional weighing from the same sample composite prepared for original test e.g. if 20 tablets were crushed to fine powder and a portion utilized for analysis, weighing from the same crushed powder and repeat the analysis. In case of dissolution and uniformity of dosage units or assay test with intact tablets, (where portion of composite powder is not tested) repeat the analysis with additional tablets collected during the original sampling and by preferably same analyst in duplicate (fresh Test preparation) for the test where OOS is observed under the supervision of senior person If same analyst is not available another competent analyst may be assigned for retesting.
In case if the OOS is observed in the test of uniformity of dosage units, Blend uniformity and Dissolution and if laboratory error is identified. Perform the retesting by same analysts (fresh Test preparation) for the test where OOS is observed. If same analyst is not available another competent analyst may be assigned for retesting.
Before initiating retesting, examine the condition and quality of sample i.e. physical description, storage environment etc. if sample is found to be homogeneous, matching as per material/product description and stored under appropriate environmental conditions, conduct retesting, If the integrity of sample can be questioned i.e. contaminated or degraded or exposed to undesired condition, exhausted, then follow the procedure specified for re- sampling and perform the retest on re-samples material/product.
Retesting may or may not be required based on the type of error occurred.
The following are examples where retesting is required.
If an error is occurred due to wrong dilution or wrong amount of sample or standard weight.
Insufficient extraction of samples or standard.
In such cases a correction in the formula as per wrong dilution or pipetting volume is not acceptable and required retesting.
The following are examples where retesting is not required.
Error occurred due to calculation, e.g. wrong dilution factor used for calculation.
Data wrongly labeled, e.g. in case of multiple strength analysis, batch number wrongly interchanged and entered.
In such case retesting is not required. Correct the calculation/data recalculate the test results and document the same. In-validate the earlier results as specified above and report the corrected test results.
The use of retest result is only acceptable where an investigation of the failure reveal that there has been an analytical error.
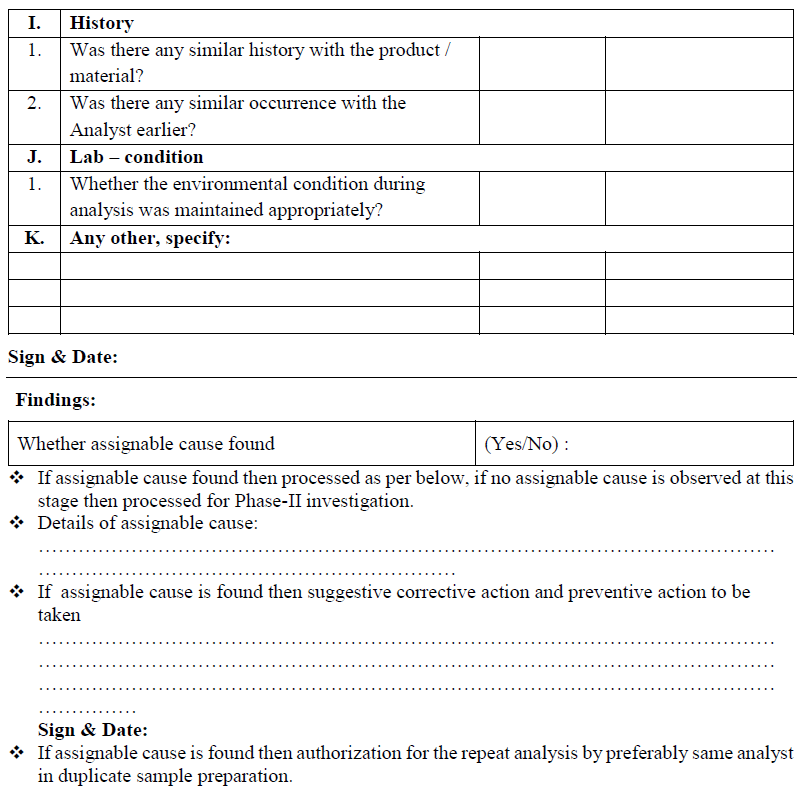
Conclusion: Based on investigation QC head or his designee shall review the investigation report, if the retesting results are comparable, no variability between results and meet the specifications, then results of retesting shall be averaged and reported as final value and submit the investigation report to QA department for final review.
If retesting results are not comparable and variability between the results then do not average the results and submit the report to QA department for further investigation.
QA Head/ Designee shall review the investigation report; if retesting result is satisfactory then material/product shall be release and invalidate the initial OOS result.
If the retesting result does not meet the specification, then this confirms that there was no laboratory error during the analysis and a non assignable errors confirms.
Non-assignable cause:
If assignable cause is not identified during laboratory investigation then Phase- II investigation shall be initiated.
If an OOS is observed for stability sample, follow the procedure specified below.
Investigate the cause of OOS as above.
Review the data of previous time points to confirm the OOS results obtained. Check whether trend shows the deviation (increase or decreases) of the test value from initial value which finally resulted in OOS.
Review the stability data of other batches of same strength, different strengths and packs to understand the probable cause of OOS result.
If the assay content is found to be reduced, check the concomitant increase in impurity content.
Review the degradation study data pathway and check the results of corresponding stress condition to understand the degradation pattern.
Check the container closure system and correlate the same with degradation pathway to understand the probable cause e.g. if a product is packed in a container having more head space and oxidation study reveals significant degradation, product may show increase in impurity contents at accelerated condition.
Inform to QA, production, R&D, Regulatory affairs and concerned departments for further necessary action.
Confirm the description of sample and how the sample was stored after being withdraw from the stability chamber until it was tested. This is rule out improper handling of sample in the laboratory. If deterioration due to improper storage of the sample is suspected, then re-testing on a new sample from stability chamber shall be performed to verify the accuracy of initial result.
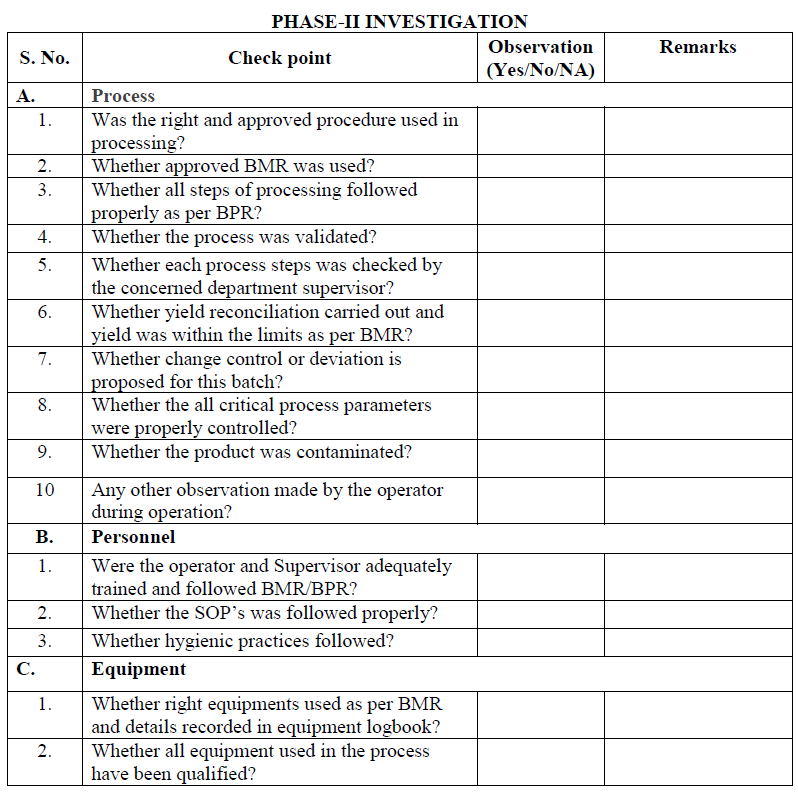
Phase-II investigation:
Phase-II investigation shall be carried out by QA Head/ Designee or his designee and production head or his designee. They may consult (if required) with warehouse, QC, and engineering and R & D department for evaluating the root cause of the OOS results reported for the sample.
The objective of full scale investigation is to identify the root cause of OOS result and take appropriate corrective and preventive action.
A full scale investigation shall include a review of production and sampling procedure and additional laboratory test.
QA department shall initiate full scale investigation. Full scale investigation may include the following investigation but not limited to:
QA Assessment:
The QA assessment (Only if deemed necessary by outcome (s) preliminary laboratory investigation i.e. No laboratory error or part of full scale investigation) shall be performed by QA personnel to determine the root cause of OOS result. Assessment may only considered if it has been determined at any point of investigation process.
QA designee shall review the following areas but not limited to:
Review of production documents.
Review of product development and validation data.
Review of product history and trends.
Review of product deviation.
Review of raw material stock register of warehouse and analytical data.
Review of Equipment/instrument validation /calibration record.
Review of sampling procedure.
Review the all documents related to other activity like utility, cleaning, sanitization, gowning procedure, personnel hygiene and environmental condition.
Shop floor investigation:
Designee of QA/QC/Production/Engineering/Warehouse shall conduct the shop floor investigation which includes review of production/general process and related procedure as per check list.
If assignable or root cause is identified during shop floor investigation then the investigation completed. However investigation must be extended to other batches of same product and other product that may be associated with failure and corrective action and preventive action shall be taken by production head and QA Head/ Designee and production head shall responsible to implementing the CAPA as per the finding of investigation.
After conclusion and QA Head/Designee decision, batch shall be Rejected/Dispositioned.
If cause of OOS result does not found during shop floor investigation then next course of action (extended additional investigation) shall be decided by QA Head/Designee.
Additional Laboratory Testing:
A full-scale OOS investigation may include additional laboratory testing. A number of practices are used during the laboratory phase of an investigation. These include retesting a portion of original sample and re-sampling.
Retesting:
Re-testing in case of raw material (only if deemed necessary by outcome(s) preliminary laboratory investigation i.e. No laboratory error) shall be performed to determine the root cause and confirm the OOS results.
Re-testing in case of in-process ,finished product and stability sample (only if deemed necessary by outcome(s) preliminary laboratory investigation i.e. No laboratory error or as a part of full scale investigation)
Re-testing shall be performed by original homogenous sample that was initially collected from the lot or batch that yielded the OOS result or resample, by two analysts in triplicate (fresh test preparation) for the test where OOS is observed (who had performed the original testing shall be preferable selected as one of analyst and another as second analyst) If same analyst is not available another competent analyst may be assigned for retesting.
Based on investigation QC head or his designee shall review the investigation report, if the retesting results are comparable, no variability between results and meet the specifications, then results of retesting shall be averaged and reported as final value and submit the investigation report to QA department for final review.
QA Head/ Designee review the investigation report if retesting result is satisfactory then material/product shall be release.
In case of raw material if retest result confirm the OOS result then material shall be rejected and QA shall inform to vendor about rejection of material and seek the root cause investigation and CAPA as applicable.
The original OOS result can only be consider invalid if the original OOS result has been determined , not be representative of material being tested based on scientifically supported conclusion of a through root cause investigation.
Release of any material can be considered if the retesting results are comparable, no variability between results and meet the specification.
Sampling error investigation:
Sampling error investigation (only If deemed necessary by outcome (s) of preliminary laboratory investigation i.e. No laboratory error or part of full scale investigation) may be performed to determine the root cause of OOS result.
QA/QC designee shall take decision to determine the sampling error as per check list.
If required re-sampling shall be performed after authorization from Quality Assurance.
Re-sampling is not considered an automatic next step of investigation process even the assignable or root cause is not found as part of laboratory preliminary investigation.
Re-sampling shall be performed by same qualified, personnel and procedure that were used for initial sample. However if investigation determines that initial sampling procedure was inherently inadequate, a new sampling procedure must be developed , documented, reviewed and approved by QA Head/ Designee.
Retest, the resample material as per assignable cause.
Retesting and re-sampling may only be considered if it has been determined at any point of the investigation reveal that:
Original sample was not representative samples.
Original sample was not stored appropriately.
Original sample was improperly sampled.
Original sample may have been adversely affected by exposure to humidity, light or heat.
If quantity available is insufficient then a resampling shall be performed (for stability sample, sample from stock sample shall be used.)
The resample/additional sample shall be collected as per requirement & shall be used for testing. The double the amount shall be retained as the “Control Sample” (wherever applicable).
On completion of resampling/additional sampling analysis, initial sample shall be destroyed.
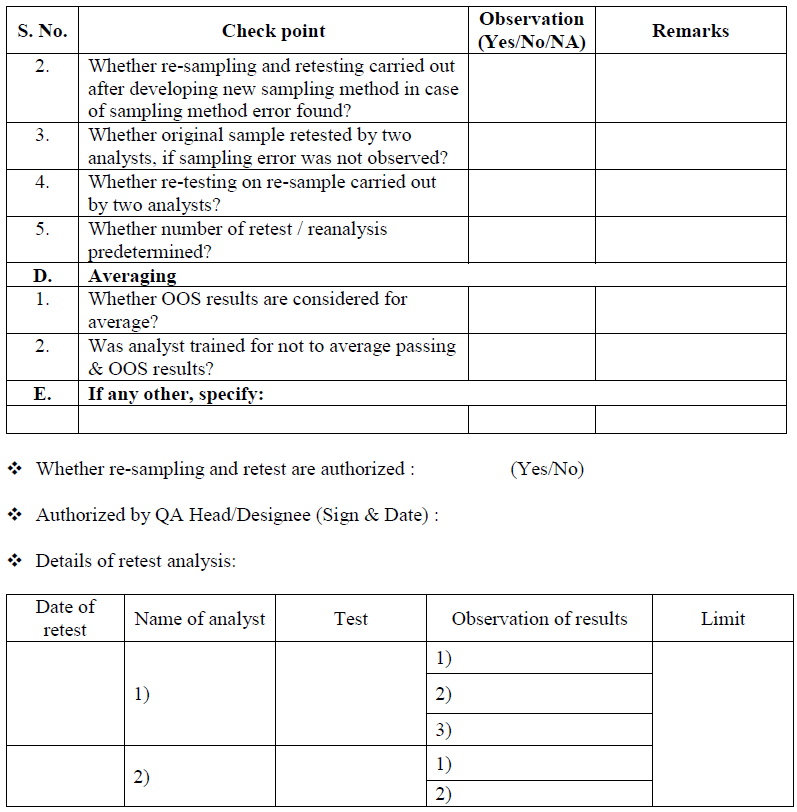
Reporting of test results:
Do not average OOS result and passing test values to obtain a passing test result. In case where a series of test results( to produce a single reportable result) are required by test procedure and some of individual results are OOS, some are within limit specification, and all are within known variability of the method, in such case provide the individual test results for evaluation and consideration by Quality assurance.
When additional testing (retest) is performed during an OOS investigation, and results of original test (OOS result) and additional retest or resample result obtained during OOS investigation is not appropriate then averaging of result should not be done because it hides variability among the individual results in such case, retain both the results.
Averaging cannot used in cases when testing is intended to measure variability within product such as powder blend/mixture uniformity or dosage form content uniformity.
If retesting results are comparable, no variability between results and meet the specifications, then results shall be averaged and reported as final value.
Batch can be release after evaluation of all individual results by Head QA/Designee. % RSD of replicates shall be NMT 2.0% in case of assay. In case of RS, all result shall be comply the specifications of respective material/products.
Hypothesis Testing (Applicable to Phase I and Phase II):
If no assignable cause that could explain in the results can be identified during the phase I and Phase II investigation protocol base study for hypothesis testing to be performed to support potential root causes if required.
Perform the impact assessment on other batches analysed in same sequence during hypothesis testing study based on protocol if required.
Interpretation of investigation results:
The complete investigation details (including OOS result retest result (if any) shall be properly interpreted to evaluate the batch and reach a decision regarding release, rejection or Disposition of the batch.
In those cases, where an investigation has revealed a cause and the suspect result is invalidated; the result shall not be used to evaluate the quality of the batch. Invalidation of a discrete test result may be done only upon the observation and documentation of a test event that can reasonably be determined to have caused the OOS result.
In those cases where the investigation indicates an OOS result is caused by a factor affecting the batch quality (i.e. an OOS result is confirmed) the result shall be used in evaluating the quality of batch. A confirmed OOS result indicates that the batch does not meet established standards or specification and shall result in batch rejection and proper disposition.
For inclusive investigations in cases where an investigation (1) does not reveal a cause for the OOS result and (2) does not confirm the OOS result, the OOS result shall be given full consideration in batch disposition decision.
Field Alert Report:
If OOS is observed in the distributed batches, a complete investigation and conclusion details shall be written in the form of field alert report within three working days and it shall be submitted to the concerned regulatory authorities.
Compilation of OOS Investigation:
All the investigation details in phase-I & phase-II, shall be compiled and summary shall be prepared.
During compilation, the impact of OOS on the following aspects shall be investigated and written in summary report:
Other manufacturing batches of same product
On-going stability
Validated procedure
Analytical testing procedure
Documentation
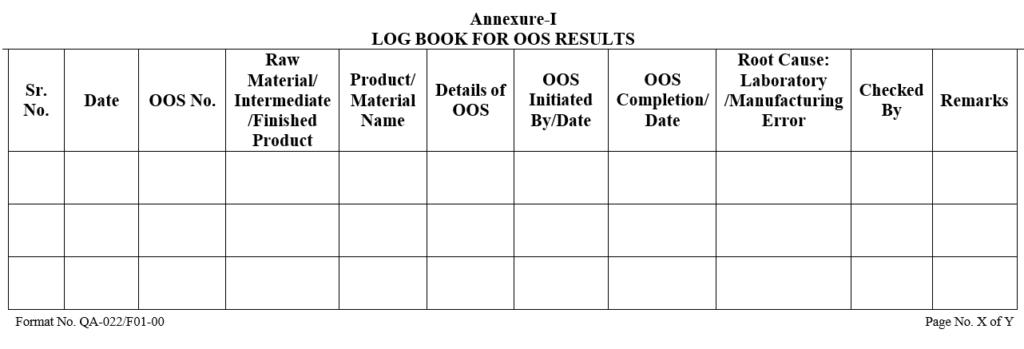
Each OOS form shall be numbered and OOS log shall be maintained by QA, as per the format.
OOS Report Number shall be consist 10 alphanumeric characters i.e. OOS/ YY-NNN, where:
First three characters “OOS” stand for “Out of Specification”.
4th character (/) is a forward slash.
The 5th and 6th characters “YY” refer to the last two digits of calendar year.(i.e. 24 for 2024)
7th character is a hyphen.
The last three characters “NNN” denote the sequential serial number of OOS (i.e. 001, 002, 003….999) for respective calendar year.
For example, OOS/24-001 refers to the first Out of Specification test result investigation report in year 2024.
Conclusion
An OOS result does not necessarily mean the subject batch fails and must be rejected.
The OOS result shall be investigated and the findings of the investigation including retest results shall interpreted to evaluate the batch and reach decision regarding release, rejection or Disposition.
A confirmed OOS result indicates that the batch does not meet established standards (or) specifications.
Trend of OOS
OOS trend shall be prepared and reviewed on half yearly basis.
Content of Trend shall be cover at least as below:
Objective
Scope
Total Number of OOS during the review period with closure status.
Month wise review of OOS during the review period.
Review of OOS based on the classification i.e. analytical error/Manufacturing error.
Categorization of Analytical error
Dilution error
Sample preparation error
Procedural error
Contamination
Procedural non compliance
Instrument performance error
Manufacturing error
Procedural non compliance
Procedure not follow
Raw Material issue
Review of OOS for closure status with open/closed beyond time line with justification.
CAPA review for OOS.
Review of Repeated OOS.
CAPA review status for repeated OOS.
Comparison with previous trend for OOS.
Department and section/stage wise review of OOS.
Review of Root Cause wise OOS
Effective analysis tools shall be used for trend analysis (pie chart, Bar chart, Parreto analysis, any other statically shall be used) during trend analysis.
References
Center for drug evaluation and research (CDER) guideline for investigation of out of specification.
Annexure
| Annexure No. | Title of Annexure | Format Number |
| Annexure-I | Log Book for OOS Results | QA-022/F01-00 |
| Annexure-II | Flow Chart for OOS Investigation | QA-022/F02-00 |
| Annexure-III | OOS Investigation Checklist | QA-022/F03-00 |
| Annexure-IV | Summary Report of OOS Investigation | QA-022/F04-00 |
| Annexure-V | Field Alert Report | QA-022/F05-00 |
DISTRIBUTION:
| Controlled Copy No. 01 | : | Head Quality Assurance |
| Controlled Copy No. 02 | : | Head Quality Control |
| Controlled Copy No. 03 | : | Head Production |
| Controlled Copy No. 04 | : | Head Engineering |
| Controlled Copy No. 05 | : | Head Warehouse |
| Master Copy | : | Quality Assurance Department |
ABBREVIATIONS:
| SOP | : | Standard Operating Procedure |
| QA | : | Quality Assurance |
| QC | : | Quality Control |
| CAPA | : | Corrective Action & Preventive Action |
| RA | : | Regulatory Affair |
| cGMP | : | Current good manufacturing practice |
| R&D | : | Research and Development |
| CQA | : | Corporate Quality Assurance |
| NMT | : | Not more than |
| RSD | : | Relative standard deviation |
| RS | : | Relative Substance |
| BMR | : | Batch Manufacturing Record |
| BPR | : | Batch Packing Record |
| A.R.No. | : | Analytical Report Record |
| Mfg. Date | : | Manufacturing Date |
| Exp. Date | : | Expiry Date |
| API | : | Active Pharmaceutical Ingredient |
| APQR | : | Annual Product Quality Review |
REVISION HISTORY:
CHANGE HISTORY LOG
| Revision No. | Details of Changes | Reason for Change | Effective Date |
| 00 | New SOP | Not Applicable | To be record manual. |
ANNXEURE – I
ANNXEURE – II
Annexure III


Annexure – IV




Annexure – V

Frequently Asked Question ?
- Why do I need to conduct an investigation if an OOS test result is obtained?
An investigation is required by FDA regulations. - What is the purpose of an OOS investigation?
To determine the cause of the OOS result. - Who should investigate OOS?
Both pharmaceutical manufacturing companies and contract laboratories. - Which parts of the CGMP regulations apply to laboratory operations?
Part 211, subparts I (Laboratory Controls) and J (Records and Reports) - Which section of the CGMP regulations specifies that products failing to meet established standards and other relevant quality control criteria will be rejected?
Section 211.165(f). - How do I handle an OOT result?
Results that are out-of-trend (OOT) can be handled similarly to OOS investigations. - Can I ignore an OOS investigation if the rejection of a batch is based on an OOS result?
No, batch rejection does not negate the need to perform the investigation! - Why is it necessary to carry out an OOS investigation if the rejection of a batch is based on an OOS result?
To determine if the result is associated with other batches of the same drug product or other products. - Which sections of the CGMP regulations require that written records of the investigation are made, including conclusions of the investigation and follow-up?
Section 211.192. - How do I carry out a meaningful OOS investigation?
A meaningful OOS investigation should be: thorough, timely, unbiased, well-documented, and scientifically defensible. - What should the analyst do, whenever possible, before discarding the test solution?
An initial assessment of the accuracy of the laboratory’s data should be conducted. Why? In this way, any hypothesis inferring laboratory error or instrument malfunction may be tested using the same solutions. - When do I need to conduct a complete failure investigation?
If the initial assessment indicates that no errors were made in the analytical process used to obtain the data, a complete failure investigation should follow. - Who is responsible for the first course of action?
The analyst. - Who has the primary responsibility for ensuring accurate laboratory test results?
The analyst. - What should the analyst do during the testing process?
The analyst should be aware of the potential problems that can occur during the testing process, and should watch for problems that create OOS results. - Which section of the CGMP regulations requires that the analyst should ensure that only those instruments meeting established specifications are used?
211.160(b)(4). - Which section of the CGMP regulations requires that the analyst should ensure that only properly calibrated instruments are used?
211.160(b)(4). - What is the usual time requirement during the LAL-endotoxin kinetic determination?
One hour or longer. - What system suitability requirements may be considered?
Drift, noise, temperature variability, photometric absorbance variability. - Why could the optical density change calculation method generate OOS results?
There is no safe method to avoid signal drift being utilized as LAL-endotoxin reaction signal. - If signal drift is a problem, then which LAL-endotoxin kinetic curves are most likely to be affected?
Turbidimetric determinations at the lowest endotoxin quantification levels. - What should be done if unexpected results are obtained and no obvious explanation exists?
Test preparations should be retained, the analyst should inform the supervisor, and an accuracy assessment should be started immediately. - Can you list some of the obvious errors that could invalidate results?
Spilling of a sample, incomplete transfer of sample, obvious pipetting errors, bubble formation in microplate wells, dirty lab equipment. - What should be done in the case of obvious errors?
The analyst should immediately document what happened. - Is it a good idea to continue with an analysis to see what results can be obtained when an obvious error is known, and that the analysis will be invalidated at a later time for an assignable cause?
No, analysts should not knowingly continue any analysis they expect to invalidate. - What should the supervisor’s responsibilities be in the case of OOS results?
A supervisor’s assessment should be objective and timely, and free from any preconceived assumptions as to the cause of OOS results. Data should be assessed promptly to ascertain if the results may be attributed to laboratory error, or whether the results could indicate problems in the manufacturing process. - What should be included in the re-examination of an immediate assessment?
Re-examination of the actual solutions, test units, glassware, and lab equipment used in the original measurements. This will allow more credibility to be given to laboratory error theories. - What steps should be taken during the supervisor’s assessment?
(1) Review training results of the analyst to determine the analyst’s competency to carry out the analysis, (2) Review the test procedure and associated SOPs for correctness, (3) Discuss the test method with the analyst to confirm analyst knowledge of, and performance of, the correct procedure, (4) Examine raw data obtained in the analysis, including all of the analytical (measurement) signals generated during analysis, such as chromatograms and spectra, and identify anomalous or suspect information, (5) Confirm the performance of the instruments (PQ), (6) Determine that appropriate reference standards, solvents, reagents, and other solutions were used and they meet quality control specifications, (7) Evaluate the performance of the testing method to ensure that it is performing to the expected standard, based on method validation data, (8) Document and preserve evidence of this assessment. - What kind of transient equipment malfunction can occur in HPLC and how can it be resolved?
An auto-injector could inject bubbles during an operation; therefore, repeated re-injections will provide strong evidence that the problem can be attributed to the instrument, rather than the sample or its preparation. - What kind of transient equipment malfunction can occur in LAL-endotoxin kinetic testing and how can it be resolved?
Photometric measurement failure could generate a radically different signal, which can radically distort the OD limits, which in turn would provide a false reaction time for the endotoxin calculation; consequently, removal of the outlier OD reading can provide strong evidence that the problem should be attributed to the instrument, rather than the sample or its preparation. - How frequently is it reasonable to experience a lab error?
Laboratory error should be relatively rare. Frequent laboratory errors could mean one or more of the following: (1) inadequate training of the analysts, (2) poorly maintained equipment, (3) improperly calibrated equipment, (4) careless work. - What should be done with OOS results if clear evidence of laboratory error exists?
The OOS test results should be invalidated. - What should be done with OOS results when evidence of laboratory error remains unclear?
A failure investigation should be conducted to determine what caused the unexpected result. - How should I select the procedure for an OOS failure investigation?
A predefined procedure should be applied. - If varying test results are obtained, and when the evidence of laboratory error remains unclear, then what may be the cause of the OOS problem?
The OOS problem may be due to either the manufacturing process or the sampling process. - Who should conduct an investigation and what departments should be included?
The QC unit should conduct the investigation, which should involve all other departments that could be implicated. - What practices are used in the laboratory phase of an OOS investigation?
(1) Retesting a portion of the original sample, (2) testing a specimen from the collection of a new sample from the batch, (3) resampling test data, (4) using outlier testing. - What conditions indicate retesting?
Situations where retesting is indicated include investigating testing instrument malfunction or identifying possible sample handling integrity problems, for example, a suspected dilution error. - Who should do the retesting?
An analyst other than the one who performed the original test should perform retesting. - Which section of the CGMP regulations requires the establishment of specifications, standards, sampling plans, test procedures, and other laboratory control mechanisms?
Section 211.160. (Note: USP 23, under General Notes and Requirements, p. 9, states: testing must be in accordance with “predetermined guidelines or sampling strategies.”) - How many times can I repeat the retests?
The number of retests to be performed on a sample should be based on company specifications contained in the relevant SOP. - What should be done if test results obtained after the predetermined number of retests are still unsatisfactory?
If, at this point, the results are unsatisfactory, the batch is suspect and must be rejected or held pending further investigation (§211.165(f)). - If I have 2 results, one original OOS result, and one of the retest results which one should be included in the batch?
In the case of a clearly identified laboratory error, the retest results would substitute for the original test results. If no laboratory or statistical errors are identified in the first test, then both test results should be reported and considered in batch release decisions. - How should I document my retest results?
In all cases the original test result should be retained, an explanation recorded. The record should be initialed and dated by the persons involved, and include a discussion of the error with supervisory comments. - How does resampling differ from retesting?
While retesting refers to analysis of the original sample, resampling involves analyzing a specimen from the collection of a new sample from the batch. (Note: testing should be done in accordance with predetermined procedures and sampling strategies (§211.165(c).) - When is resampling of the batch indicated?
A resampling of the batch should be conducted if the investigation shows that the original sample was not representative of the batch. (Note: in some cases, when all data have been examined (e.g., widely varied results obtained from several aliquots of the original composite/after determining there was no performance error made in the analysis), it may be concluded that the original sample was improperly prepared and was therefore not representative of the batch (§211.160(b)(3)). - Based on the above problems, can I use a new sampling method?
No, resampling should be performed by the same qualified, validated methods that were used for the initial sample. (Note: if the investigation determines that the sampling method was in error, a new sampling method must be developed, qualified, and documented (§211.160 and 165(c)). - What determines validity for the averaging of test data?
The validity of averaging depends upon the sample and its purpose. (Note: if the sample can be assumed to be homogeneous, i.e., an individual sample preparation designed to be homogeneous.) Using averages can provide more accurate results. For example, in the case of microbiological assays, the USP prefers the use of averages because of the innate variability of the microbiological test system. - Can you give examples when averaging can be used?
Kinetic scan of individual wells, or endotoxin data from a number of consecutive measurements (note: the determination is considered one test and one result), however, unexpected variation in replicate determinations should trigger investigation and documentation requirements (21 CFR 211.192). - Can you give examples when averaging cannot be used?
In cases when testing is intended to measure variability within the product, such as powder blend/mixture uniformity or dosage form content uniformity. - What conditions are required for the use of averaging?
(1) All test results should confirm to specifications (Note: a batch must be formulated with the intent to provide not less than 100 percent of the labeled or established amount of the active ingredient (21 CFR 211.101(a)), (2) Averaging must be specified by the test method. - Do the CGMP regulations require that the acceptance and/or rejection levels be based on statistically valid quality control criteria?
Yes, according to Section 211.165(d) of the CGMP, statistically valid quality control criteria should be used for appropriate acceptance and/or rejection levels. - How can an outlier point generate OOS results for LAL-endotoxin testing?
If OD limit calculations are used, an outlier point can generate an OD limit at the point of instrumental malfunction or noise generated. - What corrective action can you take in case of outliers?
Instrument malfunctions can occur due to power line surge, sudden lamp failure, sudden detector failure, high frequency transient noise, power failure.
55: What is an out-of-specification (OOS) result in the pharmaceutical industry?
A: An out-of-specification (OOS) result in the pharmaceutical industry refers to a test result that falls outside the established acceptance criteria or specifications for a particular product. These specifications are defined during the drug development and manufacturing process to ensure the quality, safety, and efficacy of pharmaceutical products.
56: What steps should be taken when an out-of-specification result occurs?
A: When an out-of-specification result occurs, a thorough investigation is initiated to determine the root cause and to assess the impact on product quality. The following steps are generally taken:
- Quarantine the affected batch: Prevent the distribution or use of the batch until the investigation is complete.
- Immediate retesting: Conduct a retest of the sample to confirm the initial result and rule out potential errors.
- Full investigation: Investigate the root cause, considering factors such as analytical methods, equipment, personnel, and environmental conditions. This may involve a detailed review of records, interviews, and additional testing.
- Document the investigation: Maintain comprehensive documentation of the investigation process, findings, and corrective actions taken.
- Risk assessment: Evaluate the impact of the OOS result on product quality and patient safety. Assess whether the batch should be rejected, reprocessed, or released with appropriate corrective actions.
- Corrective actions: Implement corrective actions to address the identified root cause and prevent recurrence. This may involve adjusting manufacturing processes, improving analytical methods, or enhancing training for personnel.
- Review and approval: Ensure that the investigation and corrective actions are reviewed and approved by appropriate quality assurance personnel.
- Reporting: If required, report the OOS result to regulatory authorities in accordance with regulatory requirements.
57: How can pharmaceutical companies prevent out-of-specification results?
A: To prevent out-of-specification results, pharmaceutical companies can implement the following measures:
- Robust Quality Management Systems (QMS): Establish and maintain a comprehensive QMS to ensure adherence to good manufacturing practices (GMP) and other relevant regulations.
- Validated Analytical Methods: Ensure that analytical methods used for testing are validated and suitable for their intended purpose.
- Proper Training: Provide thorough training for personnel involved in manufacturing, testing, and quality control processes.
- Equipment Calibration and Maintenance: Regularly calibrate and maintain equipment to ensure accurate and reliable results.
- Environmental Monitoring: Implement effective environmental monitoring programs to control factors that could impact product quality.
- Raw Material Control: Thoroughly inspect and control the quality of raw materials used in the manufacturing process.
- Documentation and Record Keeping: Maintain accurate and complete documentation of all manufacturing and testing activities.
- Continuous Improvement: Foster a culture of continuous improvement, encouraging ongoing reviews and updates to processes based on lessons learned.
58: What is the definition of an out-of-specification (OOS) result in pharmaceutical manufacturing?
A: An OOS result refers to a test result falling outside established acceptance criteria, indicating a deviation from the expected quality standards for a pharmaceutical product.
59: What are the primary reasons for an out-of-specification result in pharmaceutical testing?
A: OOS results can be caused by various factors, including analytical errors, equipment malfunctions, inadequate procedures, environmental factors, or issues with raw materials.
60: What immediate actions should be taken when an out-of-specification result is identified?
A: Immediate actions include quarantining the affected batch, conducting retests to confirm the result, and initiating a comprehensive investigation into the root cause.
61: How is the root cause of an out-of-specification result determined in the pharmaceutical industry?
A: Root cause analysis involves a thorough examination of factors such as analytical methods, equipment, personnel, and environmental conditions. It may include interviews, documentation reviews, and additional testing.
62: What is the role of risk assessment in handling out-of-specification results?
A: Risk assessment evaluates the impact of the OOS result on product quality and patient safety. It guides decisions on whether to reject, reprocess, or release the batch with appropriate corrective actions.
63: How can pharmaceutical companies prevent out-of-specification results in their manufacturing processes?
A: Prevention measures include implementing robust quality management systems, using validated analytical methods, providing thorough training, maintaining equipment, monitoring the environment, controlling raw materials, and fostering a culture of continuous improvement.
64: What regulatory requirements govern the reporting of out-of-specification results?
A: Reporting requirements vary by region, but pharmaceutical companies generally must report OOS results to regulatory authorities as per relevant GMP (Good Manufacturing Practice) guidelines.
65: How are corrective actions determined and implemented in response to an out-of-specification result?
A: Corrective actions are based on the identified root cause and may involve adjusting manufacturing processes, improving analytical methods, or enhancing personnel training. The actions are thoroughly documented and reviewed for effectiveness.
66: Can a batch with an out-of-specification result be released for distribution?
A: The decision to release a batch with an OOS result depends on the risk assessment. If the product’s quality and patient safety are not compromised, and appropriate corrective actions are taken, regulatory authorities may allow release.
67: How does the pharmaceutical industry ensure the ongoing improvement of processes to prevent out-of-specification results?
A: Continuous improvement involves regular reviews, updates to procedures based on lessons learned, and a commitment to addressing any identified weaknesses in the manufacturing and testing processes. It is a fundamental aspect of maintaining product quality and compliance.


