
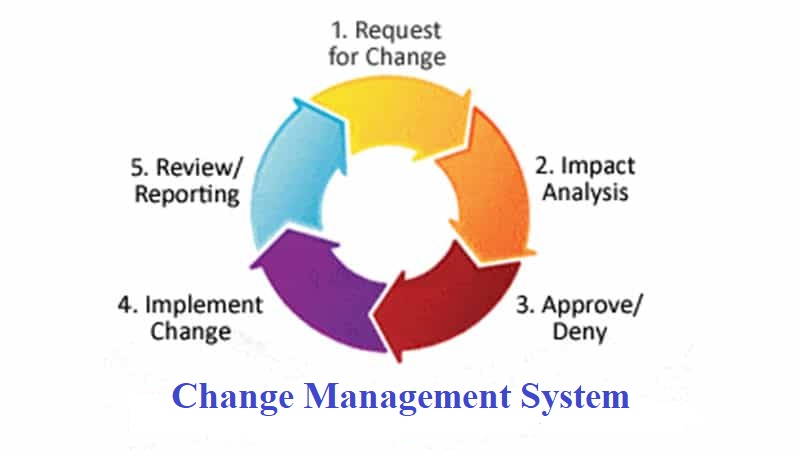
- OBJECTIVE:
To lay down a standard operating procedure for the handling of change control.
- SCOPE:
This SOP is applicable to all changes related to New product introduced at site, Facility, Equipment, Manufacturing process, Utility system, Batch size, Specifications, Analytical Procedures, documentations (SOP’s, Site Master file, Validation Master Plan, Training Manual, Safety manual, Qualification/Validation/Stability Protocols and formats etc),Container closure System, Art work of Printed packing material, Pack specifications. Changes due to changes/updation in the Pharmacopoeia, introduction of new pack size in the same manufacturing line. It shall also be applicable to changes related to Computer Systems (Software and Hardware), Item codes, BOM. Procedural changes, CAPA as outcome to investigation at {Company Name} {Company Location}.
- RESPONSIBILITY:
- Initiator: Any responsible person in the department may suggest the change and shall discuss the proposal with the section head. The section head shall be authorized to propose change and shall be responsible to:
- Initiate the Change proposal in the prescribed format along with supporting information and justification for change.
- Follow up for evaluation and decision on change(s) proposed.
- Implement the change, if approved.
- Complete the necessary activity /documentation as a result of evaluation conducted on impact assessed.
- Furnish the required information /data after implementation of change for evaluation and assessment for regularization of the proposed change.
- DEPARTMENTAL HEAD SHALL BE RESPONSIBLE TO:
- Review the Change Control format for its completeness and evaluate if the reason/ Justification given in support of the change proposed is appropriate and justified.
- Recommend the proposed change after review and the forward the same to QA.
- Ensure the implementation of the change after approval from QA.
- Review the post implementation data and forward to QA to support the closure of the change proposed.
- In case the proposed change (s) does not serve the purpose, ensure return to the original practice/position.
- QUALITY ASSURANCE SHALL BE RESPONSIBLE TO :
- To verify whether the Change Control format filled is correct and all the relevant information as necessary has been provided for evaluation.
- To evaluate the proposed change and its impact on safety, purity, efficacy of the product, cGMP, documentation and training requirements.
- Identify the actions required as part of impact assessment as consequence to the proposal made.
- Identify the departments from where evaluation of change is required and arrange to forward the proposal to the concerned department for their evaluation/comments and acceptance.
- To decide whether the Change proposed is to be forwarded for comments /evaluation and approval/rejection by Head QA, R&D or Regulatory Affairs and other functions as required.
- Monitor the implementation of proposed change, if the proposal is approved by Quality Assurance Head.
- To review post implementation information /data and arrange for closure of Change Control after verification of completion of actions as identified during the review and approval.
- QUALITY ASSURANCE HEAD/ DESIGNEE SHALL BE RESPONSIBLE TO :
- Evaluate the proposed change and to approve or reject the proposal.
- Review the post implementation data and recommend whether to regularize the proposed change or to go back to the original procedure/practice or ask for generation of additional data.
- Periodic review of Change Control status.
- OTHER DEPARTMENTS /PERSONNEL IDENTIFIED TO EXECUTE THE CHANGE SHALL BE RESPONSIBLE TO:
- Evaluate the proposed change and comment for acceptable or disapproval.
- Identify any additional activity / documents affected by the proposed change.
- Complete necessary documentation, generate data or provide justification as required.
Click the link for download word file copy of this document: https://pharmaguidehub.com/product/change-control/
- REGULATORY AFFAIRS:
- Evaluate the change proposal for any Regulatory implication, as per the filing or Registration profile and /or commitment made to the agency through Annual updates, variation application, Technical Agreement or any other form of communication.
- ACCOUNTABILITY:
- QA Head shall be Accountable for implementation of SOP.
- PROCEDURE:
- Any Changes or modification made in Facility, System, Equipment, Instrument, Process, Procedure and Documentation after initial Qualification / Validation shall be documented and facilitated through Change Control Procedure. Changes can be made in the following:
- Facility Related:
- Building Design, Lay Out (Civil layout, Man Material movement), Relocation or Replacement of Equipment, changes in Water System, HVAC & all Utilities.
- Manufacturing Process Related:
- New product introduced at site, Changes in batch size, batch formula, manufacturing steps/instructions, process parameters, In-process controls, primary packing, pack specification and pack size.
- Specification /Test Procedures Related:
- Change in the specification of Raw materials (including active, excipients, solvent, reagent, and catalysts) In-process steps, Finished Product, Change in Analytical or Microbiological test procedures (STP/GTP), Stability Protocol, Analytical Method Validation (if applicable) and any other material which may directly/indirectly affect the product quality; Changes in Packing Material specification, Art work and other printed packaging material, shall be requested to be routed through Change control process.
- Stability:
- Extension or reduction of the shelf life based on the available stability data and change in storage condition, Stability Program.
- Documentation:
- BMR & BPR, Validation Protocol, VMP, SMF, SOP, Quality manual, Safety manual, Training manual, Calibration Master Plan, logbooks related to Manufacturing QC/QA, Warehouse, Engineering and other affected documents from changes.
- Others:
- Modification/changes of item codes, Bill of material, Computer based systems, Changes proposed by marketing based on the feedback from customers, Changes proposed by Packaging Development for improving the packing /presentation of the product, changes proposed by Distribution or changes due to change in registration profiles. Changes due to updation in Pharmacopoeia, changes, due to introduction of new guidelines or cGMP requirements, Compliance issues, CAPA practices/Procedural changes.
- IDENTIFICATION OF CHANGES :
- Concerned Department personnel shall identify the changes required in the system and subsequently inform to Department Head.
- Change control for a permanent change can be raised in case of Major Deviations after three Planned Deviations.
- If the minor deviation occurs frequently, then, a trend shall be made for the deviations. Based on the trend data, a permanent change can be implemented through Change Control.
- INITIATION OF CHANGES:
- Change(s) shall proposed by section head of the department which are aimed at increasing efficiency, reducing cost, reducing manpower spent, and improving productivity, better traceability for GMP/GLP/documentation/Quality System improvement and safety related issues without impacting on the product purity and quality.
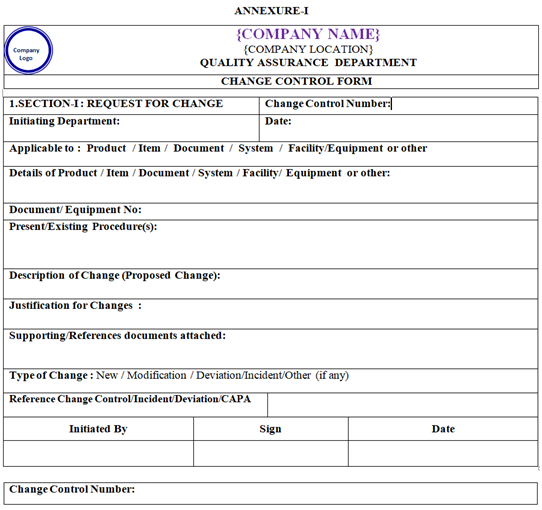
- The initiator shall initiate request for Change Control form to QA.
- QA officer/Executive shall make a photo copy of change control form and put controlled copy stamp on all pages and the sign with date on “Issued by” on first pages. As per Annexure-I.
- On issuance of the Change Control form, the following details shall be furnished by initiator in the form.
- Date of Initiation, Department.
- Description of Change: A brief outline of the proposed change.
- Justification for Change.
- List of documents attached /Reference/Justification/Rationale.
- Justification for change (may include, not limited to the following):
- Regulatory requirement based on guidelines.
- Detail of product/Item/Document/System/Facility/Equipment or other
- GMP /GLP improvement.
- Compliance to audit observation.
- Capacity enhancement.
- Expansion /renovation of the facility to ease out storage, man material movement.
- Process automation.
- Improvement suggested by marketing department.
- CAPA as taken as the outcome of the investigation conducted.
- Environmental conditions improvement.
- Personnel skill improvement.
- Trend data
- Stability data
- Annual Product review
- Process Validation, etc.
- Necessary supporting documents to justify the proposed change as applicable shall be attached with the Change Control format.
- Tentative date of completion and type of change.
Click the link for download word file copy of this document: https://pharmaguidehub.com/product/change-control/
- EVALUATION OF CHANGE :
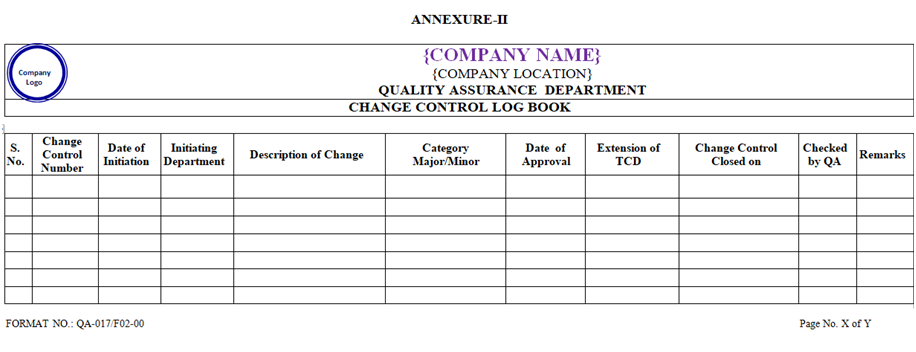
- On receipt of Change Control form QA Executive or his designee shall log the proposed change in the Change Control log book.
- Assignment of Change Control Number:
- The Change Control Number shall be assigned as XXX/CC/YY-NNN, where
| XXX | : | {Initial of Company Name} |
| CC | : | Denotes Change Control |
| YY | : | Last two digits of the Calendar Year |
| NNN | : | Indicates to three digit serial number e.g. 001, 002, 003….. |
Example : XXX/CC/23-001 Denotes first Change Control raised in year 2017
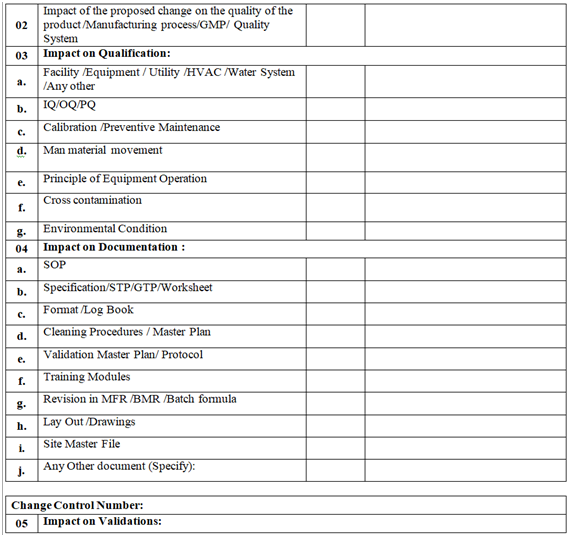
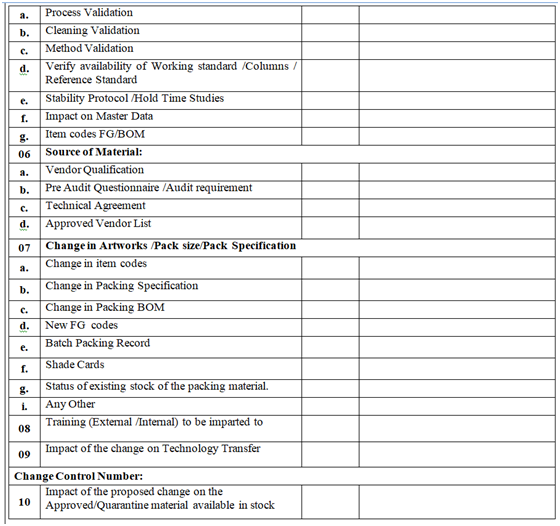
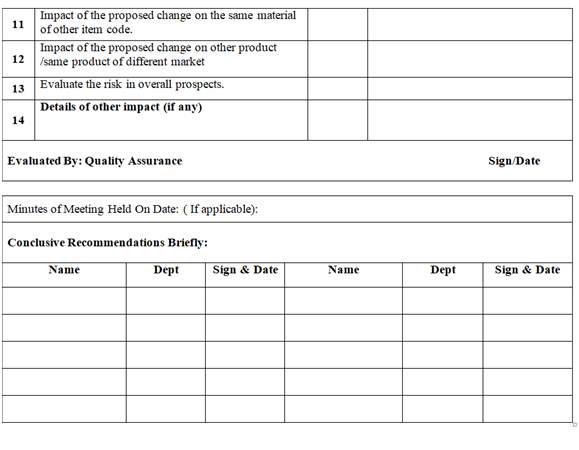
- QA shall assess the impact of the change proposal on:
- Product quality and stability
- Quality Systems /GMP
- Process /quality parameter
- Potential for mix ups, cross contamination
- Man Material movement
- Container closure system /Artwork /Pack specification /Labelling
- Validations /Qualifications
- Application to Regulatory agencies /fillings
- Computer system (Hard ware and software system Training)
- Training
- Any other procedural change which may have reflection of the proposed change.
- Initiator department shall assess the impact evaluation of change and reference of impacted documents shall be mentioned in section-II of change control form
- Initiating department head shall evaluate the change proposed and give his/her approval for the change
- The impact of the proposed changes on documents as listed shall also be done (but not limited to):
- MFR/BMR/BPR
- SOP
- BOM/Item codes
- Specification/GTP/STP/Worksheet
- Validation/Qualification Protocol/Validation Master Plan/Qualification Master Plan/Cleaning Master Plan/Analytical Method validation
- Stability Program /Protocol
- Formats of the documentation
- Drawing (s), facility layouts
- Technical Agreement (s)
- Site Master File
- Log books/records
- Any other, such as change in batch number coding system, reference sample from Marketing.
- As part of the evaluation of the proposed change, identification for the actions as listed below shall also be evaluate
- Validation Trial/Experimental Batch Manufacturing
- Qualification (IQ/OQ/PQ)
- Charging product on stability
- Need of training
- Development of any new procedure /method
- Prior Approval/Information/ Notification to Regulatory Body/ Business Partner/Key customer (s)/internal information to other Department.
- During review QA Executive / designee shall request any further information/data supporting the proposed change from initiator if required.
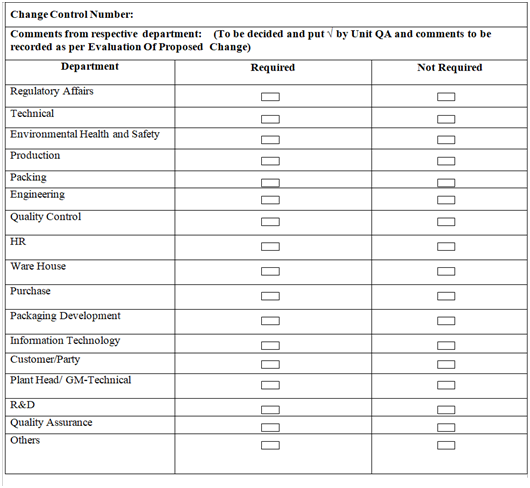
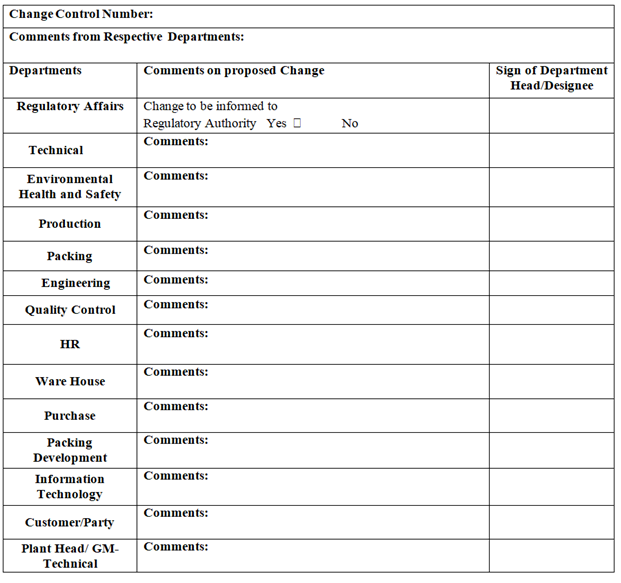
- Based on the review, QA Executive / designee shall identify the department /personnel whose comments /opinion or evaluation is required for the proposed change, based on the nature of change. This may include, but not limited to Production, Quality Control, Warehouse, Purchase, R&D, Marketing, Engineering, Regulatory Affairs or any other as applicable.
- QA Executive / designee shall forward Change Control for getting evaluation by the identified internal departments /personnel.
- After completion of the evaluation by all concerned initiator shall forward the Change Control to QA for approval.
- In case the change proposed, requires evaluation / approval from R&D, Regulatory Affairs, or any other department or customer which is not operating at the site, the site. QA shall send the scanned copy of the change control to the relevant department. After evaluation, that department shall give his comment, acceptance or rejection on the format and shall send same back to the respective site.
- Regulatory department shall evaluate the proposed change and shall identify the requirement for the following, prior to implementation of change by considering the regulatory implication, if required:
- Notification of change to customer /Agency
- Obtaining approval of change by customer
- Regulatory filing for any variation.
Click the link for download word file copy of this document: https://pharmaguidehub.com/product/change-control/
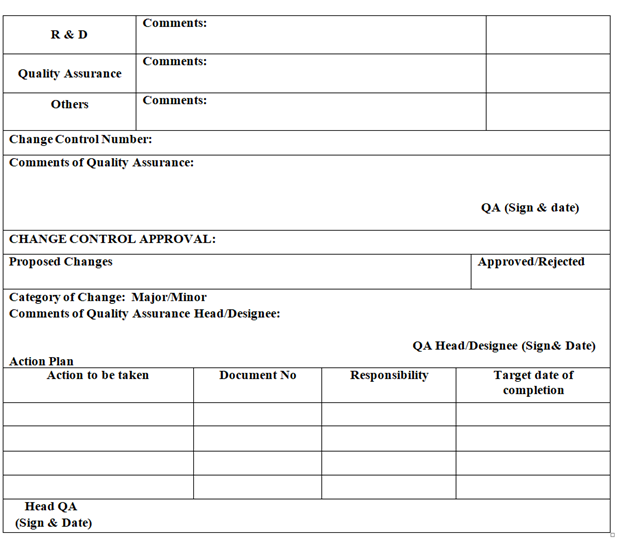
- APPROVAL OF CHANGE :
- Based on the comments and evaluation done by Executive QA / designee, Quality Assurance Manager or his designee shall:
- Give decision for approval /rejection on the change proposal.
- Classify the change as Critical/Major/Minor (if the proposed change is approved).
- Head QA shall assigns level of change as:
- Major: Changes which may have a direct and moderate impact upon the identity, quality, purity, potency, strength, stability, safety, efficacy or physical characteristic of the product such as:
- Change related to process parameters.
- Introduction of new API / new product.
- Change in software of equipment’s.
- Stability program or shelf life of the product
- Changes in environmental conditions.
- Change in cleaning procedure and cleaning agent
- Change in product usage directions
- Changes in formula, source of API and starting material.
- Changes related to the process/ process parameters.
- Changes in primary packing material.
- Addition or replacement of equipment
- Any modification in the facility, Utility systems
- Changes related to specifications, Analytical methods.
- Change in Batch Size.
- Finalization of yield limit.
- Minor: Changes which do not have direct impact upon the identity, quality, purity, potency, strength, stability, safety, efficacy or physical characteristic of the product such as:
- Changes in the recording formats.
- Correction of typographical error in documents.
- Change in secondary and tertiary packing material.
- Revision of SOPs.
- Change in Site Master File.
- Addition or replacement of equipment
- Any modification in the facility, Utility systems
- Changes related to specifications, Analytical methods.
- Change in Batch Size.
- Finalization of yield limit.
- Above changes will be considered in minor if they have not direct and moderate impact upon the identity, quality, purity, potency, strength, stability, safety, efficacy or physical characteristic of the product.
- In case of disagreements on decision for recommending the change among identified evaluators, the final decision shall be taken by Quality Assurance Head with justification.
- In case the change is not approved, Quality Assurance Head or designee shall give reason for not approving the proposal.
- The same information shall be updated in the log book and will be given to Department heads from where the proposal was initiated. Head QA shall list out actions to be taken, responsible person for the task and Target date of completion of the task.
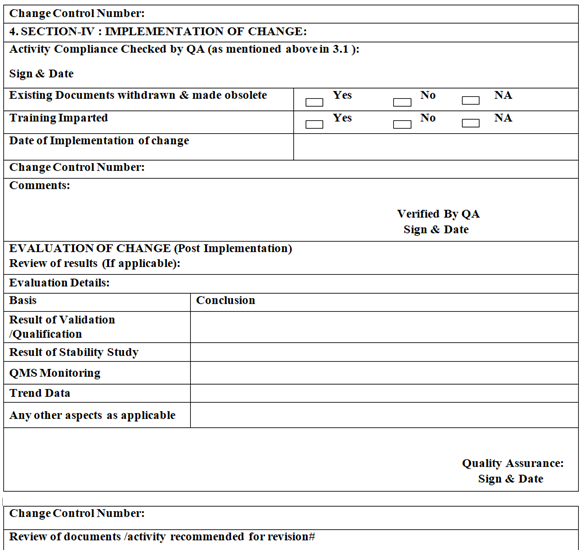
- IMPLEMENTATION OF CHANGE:
- QA Executive / designee shall communicate the same to initiator to implement the approved change.
- Initiator shall refer the details of instructions /comments /actions /documents as advised by QA in Change Control format.
- The relevant documents shall be revised and approved change shall be implemented.
- Departmental head shall ensure the implementation as approved by Quality Assurance head.
- In case of delay, tentative date of implementation of proposed changes shall be extended with justification and approved by Head QA.
- POST IMPLEMENTATION EVALUATION:
- Based on the change control proposal, Head QA shall decide the requirement for post implementation evaluation.
- In case post implementation evaluation is required Head QA shall document his decision in the change control form and action to be taken as a part of post implementation evaluation i.e., documents to be review, trend to be review, stability monitoring, etc.
- In case post implementation evaluation is not required, Head QA shall document his decision in the change control form and provide justification for the same.
- The initiator department shall submit all supporting data/information to QA for evaluation.
- If the data /information post implementation is found satisfactory, QA shall authorise for regularisation of the change. In case the data /information are not satisfactory QA shall advise to return to the original /practice and discontinuation of the approved change.
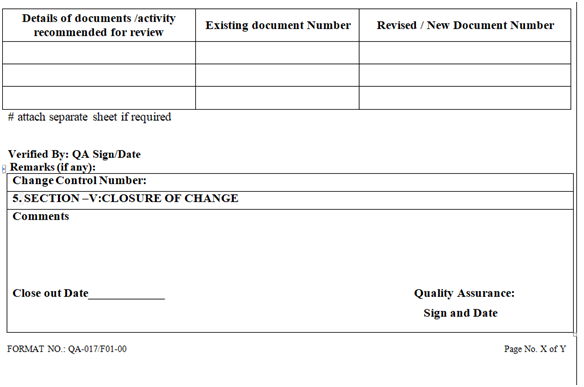
- CLOSURE OF CHANGE CONTROL
- After the post implementation evaluation of change implemented, QA Executive / designee shall do the closure of the change control form after.
- Ensuring results and supporting data.
- Completion of the required activities /documents identified during the evaluation and approval of change.
- Periodic quarterly review shall be performed by QA during March, June, September and December of each year to check for the closure of the Change Control Form, which is waiting for closure. Status report shall be prepared by QA as per “Annexure-III.
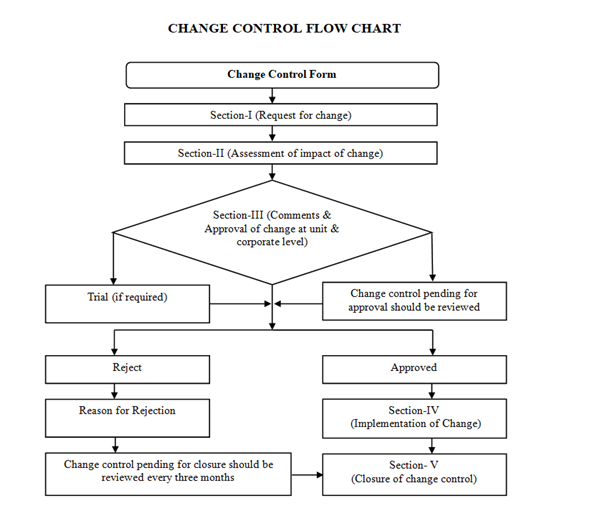
- The change control procedure shall be followed as per the flow chart:
- All activities pertaining to the implementation of change control shall be completed with 180 working days from the date of approval of change control. If activity is not completed then extension can be granted for maximum 90 days. Maximum 2 extensions can be taken. If after 2 extensions task is not closed then justification head QA can reject the change control.
- TRENDING OF CHANGE CONTROL:
- Tracking and trending of change control shall be carried out by QA.
- An Excel file shall be generated and used for logging all change control. Trend Evolution shall be done based on the following criteria:
- Review of change control based on their classification.
- Review of change control based on their closure status.
- Review of change control based on their quality Impact / Not Impact.
- Review of change control based on repeatability.
- Department wise review of change control
- Review change control based on Extension.
- Such trend shall be prepared & reviewed by QA on every six months. If less than three change control will raised in six months then trend shall be prepared & reviewed by QA on annually basis.
- If any change control shared with customer via Mail communication for change approval then customer shall be approved the changes within 10 working days of {Company Name}. The 10 working days shall be counted from the date of mail communication if customer will not give any response within stipulated time period then it will be considered as approved change control from customer side.
Click the link for download word file copy of this document: https://pharmaguidehub.com/product/change-control/
- REFERENCES:
- Not Applicable
- ANNEXURES:
| ANNEXURE No. | TITLE OF ANNEXURE | FORMAT No. |
| Annexure – I | Change Control Form | QA-017/F01-00 |
| Annexure – II | Change Control Log Book | QA-017/F02-00 |
| Annexure – III | Status of change control pending for closure | QA-017/F03-00 |
| Annexure – IV | Justification for delay | QA-017/F04-00 |
| Annexure – V | Change Control Trend Analysis | QA-017/F05-00 |
| Annexure – VI | Action plan for change control based on trends | QA-017/F06-00 |
ENCLOSURES: SOP Training Record.
DISTRIBUTION:
- Controlled Copy No. 01 : Head Quality Assurance
- Controlled Copy No. 02 : Head Production
- Controlled Copy No. 03 : Head Quality control
- Controlled Copy No. 04 : Head Engineering
- Controlled Copy No. 05 : Head Warehouse
- Controlled Copy No. 06 : Head Human Resources
- Master Copy : Quality Assurance Department
ABBREVIATIONS:
| SOP | : | Standard Operating Procedure |
| Ltd. | : | Limited |
| No. | : | Number |
| BMR | : | Batch Manufacturing Record |
| BPR | : | Batch Packing Record |
| MFR | : | Master Formula Record |
| QA | : | Quality Assurance |
| QC | : | Quality Control |
| HVAC | : | Heating Ventilation and Air Conditioning |
| VMP | : | Validation Master Plan |
| SMF | : | Site Master File |
| F & D | : | Formulation and Development |
| RM | : | Raw Material |
| IP | : | In process Material |
| FP | : | Finished Product |
| PM | : | Packing Material |
| BSE | : | Bovine Spongiform Encephalopathy |
| TSE | : | Transmissible Spongiform Encephalopathy |
| GMP | : | Good Manufacturing Practices |
| STP | : | Standard Testing Procedure |
| AMV | : | Analytical Method Validation |
| DMF | : | Drug Master File |
| COS | : | Certificate of Supplier |
- REVISION HISTORY:
CHANGE HISTORY LOG
| Revision No. | Details of Changes | Reason for Change | Effective Date |
| 00 | New SOP | Not Applicable | To be filled Manual |
ANNEXURE-I

Click the link for download word file copy of this document: https://pharmaguidehub.com/product/change-control/
ANNEXURE-II
ANNEXURE – III
ANNEXURE – IV

ANNEXURE – V

ANNEXURE – VI

Click the link for download word file copy of this document: https://pharmaguidehub.com/product/change-control/



